Random Forest Tutorial
Author: Gavin Douglas
First Created: 7 March 2017
- Introduction
- Background
- Load Packages and Read in Data
- Pre-processing
- Assessing Model Fit
- Identifying Important Features
This tutorial is intended to teach beginners the basics of running random forest (RF) models on microbial sequencing data. Users may also be interested in MetAML, which implements RF along with other machine learning techniques with a simple workflow for metagenomic data. Importantly, the below commands are not the best practices for all datasets. An important part of data analysis is determining what approaches would be best for your particular problem. However, as shall be described below, it's easy to create biased models if you select parameters based on observations you made while exploring your data.
Often when running machine learning tools you would split up your samples into a training set (often ~70%) to build your model and a test set (often ~30%) to evaluate your model. Partitioning allows an estimate of the model performance to be inferred on different data than was used to train the model. When sample sizes are large enough researchers sometimes create a third partition, the validation set, which is used to select (or tune) parameters for the model. However, often in microbial datasets sample sizes are small (e.g. often in the range of 10-50 samples), in which case alternative approaches need to be taken. Cross-validation, where different subsets of your data are systematically split into training and test sets to estimate model performance, is a family of approaches to get around this problem (a few examples are shown in this blog post). Often this actually isn't necessary for RF models given the internal measure of out-of-bag error which is calculated for each model (described below).
The below tutorial will focus on a dataset of 40 samples, where partitioning the data into training and test sets would not be helpful. Operational taxonomic unit (OTU) relative abundances are the features of this dataset. All samples have inflammation scores (IS) associated with them (ranging from 0-1). Samples with IS >= 0.5 were grouped as inflamed, while those with a value < 0.5 were put in the control group. This means that each sample has an inflammation category and a quantitative value associated with it, which will allow us to try running RF classification and regression models using the same features. Note that this example dataset was simulated for this tutorial.
You will need the below data and R packages to run all the commands in this tutorial. I have also noted the version of each package that I was using for this tutorial. You shouldn't need to use exactly the same versions as I did, but be aware that there could be updates to these packages since this tutorial was posted.
- Tutorial OTU table and mapfile.
- R (v3.3.2)
- RStudio - recommended, but not necessary (v1.0.136)
- randomForest R package (v4.6-12)
- rfUtilities R package (v2.0-0)
- caret R package (v6.0-73)
- plyr R package (v1.8.4)
- e1071 R package
- Original RF paper: Leo Breiman. 2001. Random Forests. Machine Learning 45(1):5-32.
- Paper describing the randomForest R package (includes a great description of the algorithm and helpful examples): Andy Liaw A and Matthew Wiener. 2002. Classification and Regression by randomForest. R News 2(3):18-22.
- Reference for permutation test to test RF model significance: Melanie A. Murphy et al. 2010. Quantifying Bufo boreas connectivity in Yellowstone National Park with landscape genetics. Ecology 91(1): 252-261.
- scikit-learn: a set of useful tools and workflows for running machine learning in Python.
- MetAML: a Python package based on scikit-learn produced by the Segata Lab from Trento University for running machine learning on metagenomics datasets. They have written a tutorial and research paper for this tool.
- caret: an R package that wraps many machine learning and other predictive tools. There are many online tutorials on how to use caret to run RF models, such as this short example. The coursera course Practical Machine Learning focuses on this package.
RF models are run when researchers are interested in classifying samples into categories (called classes) of interest based upon many different variables, such as taxa, functional categories, etc (called features). RF can also be used to regress features against quantitative data (e.g. height in cm rather than splitting samples into categorical groups like short and tall).
RF models are based on decision trees, which can be used for classification of a discrete variable or regression of a continuous variable. The term Classification and Regression Tree (CART) is often used to describe these decision trees. The wikipedia article on RF describes how this method contrasts with other CART methods.
Briefly, the RF algorithm involves randomly subsetting samples from your dataset and building a decision tree based on these samples. At each node in the tree mtry (a set parameter) number of features are selected from the set of all features. The feature that provides the best split (given any preceding nodes) is chosen and then the procedure is repeated. This algorithm is run on a large number of trees, based on different sample subsets, which means that this method is less prone to overfitting than other CART methods.
Since each decision tree in the forest is only based on a subset of samples, each tree's performance can be evaluated on the left out samples. When this validation is performed on all samples and trees in a RF the resulting metric is called the out-of-bag error. The advantage of using this metric is that it removes the need for a test set to evaluate the performance of your model.
Also, the out-of-bag error can be used to calculate the variable importance of all the features in the model. Variable importance is usually calculated by re-running the RF with one feature's values scrambled across all samples. This difference in accuracy between this model with the scrambled feature and the original model is one measure of variable importance.
Sometimes RF is run to perform feature selection on a dataset. This can be useful when there are thousands of features and you'd like to reduce the number to a less complex subset. However, it's important to realize that you need to validate selected features on independent data. Click here to see a simple demonstration for why this is important.
Once you're in the R environment you'll need to load the required R packages (which have a number of dependencies as well):
library("randomForest")
library("plyr") # for the "arrange" function
library("rfUtilities") # to test model significance
library("caret") # to get leave-one-out cross-validation accuracies and also contains the nearZeroVar function
Often setting your working environment simplifies working in R:
setwd("/Path/to/my/RF_tutorial/")
After downloading the tutorial OTU table and map file from the above links you can read them into R:
otu_table <- read.table("otu_table_RF_tutorial.txt", sep="\t", header=T, row.names=1, stringsAsFactors=FALSE, comment.char="")
metadata <- read.table("metadata_RF_tutorial.txt", sep="\t", header=T, row.names=1, stringsAsFactors=TRUE, comment.char="")
Now that we have the data read in R it's a good idea to explore the data a bit. A couple of basic things that we can do is double-check that the object has the expected number of rows and columns ("dim" returns the number of rows first followed by the number of columns):
dim(otu_table)
[1] 1000 40
dim(metadata)
[1] 40 2
So there are 40 samples in each file, 1000 rows in the OTU table (each corresponding to a different OTU), and 2 columns of metadata.
It's also a good idea to look at summaries of your data. This is a little more difficult for the OTU table since there are so many columns, but it's easy for the metadata file (using the "str" and "summary" functions):
str(metadata)
'data.frame': 40 obs. of 2 variables:
$ IS : num 0.76 0.71 0.86 0.72 0.53 0.96 0.97 0.85 0.66 0.55 ...
$ state: Factor w/ 2 levels "control","inflamed": 2 2 2 2 2 2 2 2 2 2 ...
summary(metadata)
IS state
Min. :0.0100 control :20
1st Qu.:0.2175 inflamed:20
Median :0.4950
Mean :0.5012
3rd Qu.:0.7625
Max. :0.9700
These steps are important to make sure you're working with the right data and that it's in the format you're expecting. However, it's also important to avoid over-exploring your data so that you don't bias your analyses.
Pre-processing is one of the most important steps in machine learning. This is because often the difference between the best performing model isn't the choice of algorithm, but simply how the data is pre-processed. RF doesn't make any assumptions about how the data is distributed so it often is not necessary to transform your data. However, reducing noise in your input data will improve model performance. An easy way to do this is to throw out features that are rare or have very low variance across samples. Any cut-offs used at this step are slightly arbitrary and would depend on the dataset.
A basic way to decide which features are unlikely to be informative is if they are non-zero in only a small number of samples. It's a good idea to take a look at this distribution for all features to help choose a reasonable cut-off.
To quickly figure out and plot how many non-zero values each OTU has:
otu_nonzero_counts <- apply(otu_table, 1, function(y) sum(length(which(y > 0))))
hist(otu_nonzero_counts, breaks=100, col="grey", main="", ylab="Number of OTUs", xlab="Number of Non-Zero Values")
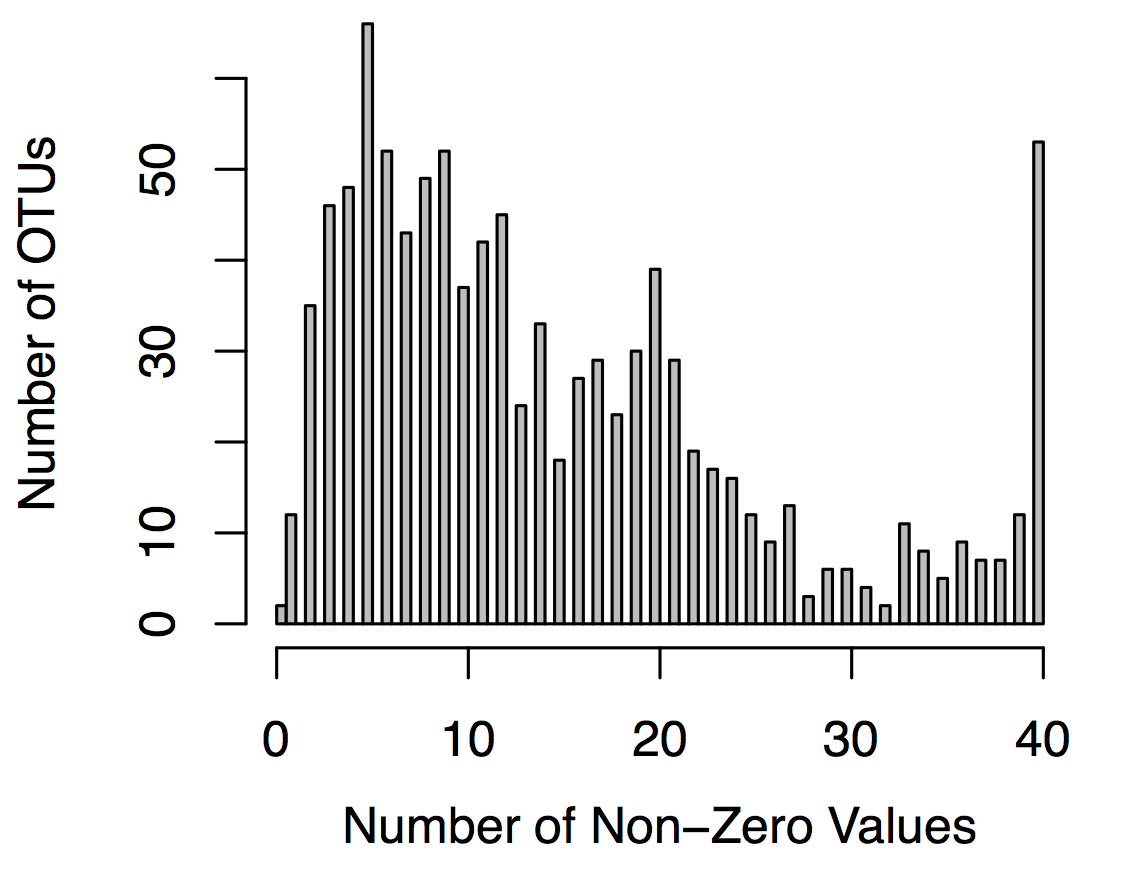
This histogram shows a typical distributions for OTU tables: most OTUs are rare and found in only a few samples. These OTUs are less likely to help our model so we can throw them out. Typically researchers discard OTUs that are zero in greater than 75-90% of samples although these cut-offs are somewhat arbitrary. This cut-off could be optimized if you had an independent dataset or partition of your data that you could use as a validation set.
This R function removes features that are non-zero less than a specified proportion of the time:
remove_rare <- function( table , cutoff_pro ) {
row2keep <- c()
cutoff <- ceiling( cutoff_pro * ncol(table) )
for ( i in 1:nrow(table) ) {
row_nonzero <- length( which( table[ i , ] > 0 ) )
if ( row_nonzero > cutoff ) {
row2keep <- c( row2keep , i)
}
}
return( table [ row2keep , , drop=F ])
}
Based on the above histogram I removed OTUs that have non-zero values in <= 20% of samples:
otu_table_rare_removed <- remove_rare(table=otu_table, cutoff_pro=0.2)
How many OTUs are left?
dim(otu_table_rare_removed)
[1] 647 40
647 OTUs are remaining, which means that 35% of the OTUs were excluded.
To re-normalize_table so that each sample's column sums to 100 (you can use the ColSums function afterwards for a sanity check):
otu_table_rare_removed_norm <- sweep(otu_table_rare_removed, 2, colSums(otu_table_rare_removed) , '/')*100
We might also be interested in excluding OTUs that don't vary much across samples. This usually isn't different from excluding OTUs based on their number of non-zero values. However, this can be useful for other types of feature tables. You may be interested in trying out the nearZeroVar() function from the caret R package to perform this filtering.
Random forest doesn't make any assumptions about how your input features are distributed so in many cases it isn't necessary to transform your data. However, certain transformations that help make features more comparable across samples can be useful when using relative abundance data. Note that the below discussion of data transformations is only applicable to microbiome sequencing data and does not represent standard approaches for transforming data in general prior to Random Forest.
This can be important to do to help resolve the issue of compositionality inherit in sequencing datasets. Because the data is compositional that can make it very difficult to compare relative abundances across samples. For example, if a feature is at 90% in sample A and missing in all others then the relative abundance of all other features in sample A will be lower on average compared to the other samples (regardless of what the actual absolute abundances in each sample are). There are many possible ways to transform microbiome sequencing data and unfortunately it usually is not clear which method would be most appropriate for machine learning. For further discussion on this issue you can see here and here.
One approach is to standardize the data by subtracting each sample's mean (center) and then dividing by the sample's standard deviation (scale). In other words, each value is converted into a Z-score. This is easy to do in R:
otu_table_scaled <- scale(otu_table_rare_removed_norm, center = TRUE, scale = TRUE)
Another method is to take the inverse hyperbolic sine and then to mean center by sample, which is also straight-forward to run in R:
otu_table_asinh_mean_centred <- scale(asinh(otu_table), center=TRUE, scale=FALSE)
In this case I will use the standardized data, but it should be clear that this is not the best transformation for all datasets. You should think carefully about what transformation is most likely to help your model's performance without detracting too much from the interpretability.
Another transformation you should be familiar with for microbiome sequencing data in the centred log-ratio (CLR) transformation. This is one of the most common transformations for read counts in each sample. Note that again typically this is performed on read counts rather than relative abundances, because a pseudocount needs to be added to deal with 0 values in the table. A pseudocount choice of 0.5 or 1 is typically chosen when dealing with read counts, although other approaches, such as those implemented in the zCompositions R package, are also available. This transformation can be easily calculated with the clr function in the compositions R package.
Now that we're content with our pre-processing steps, we need to get the input tables into the correct format (note that the metadata column will be the last column of each input dataframe).
To prep input tables for classification of state:
otu_table_scaled_state <- data.frame(t(otu_table_scaled))
otu_table_scaled_state$state <- metadata[rownames(otu_table_scaled_state), "state"]
To prep input tables for regression of inflammation score (IS):
otu_table_scaled_IS <- data.frame(t(otu_table_scaled))
otu_table_scaled_IS$IS <- metadata[rownames(otu_table_scaled_IS), "IS"]
As described above, the 2 parameters for a RF model are the number of trees in the forest (ntree) and the number of features randomly sampled at each node in a tree (mtry). The more trees you run in your forest the better the model will converge. Below I used 501, which is similar to the default, but in practice you may want to use something like 10,001 trees for a robust model (depending on the computational time). Note that I usually choose odd numbers of trees to ensure there are never any ties for binary classification models. Unless you have a reason to change mtry beforehand (or can optimize it with an independent partition of data) it's better to use the default values. The default mtry values differ for RF classification and regression - as shown in the below plot.

Finally, before running the models you should set the random seed so that they will be reproducible.
set.seed(151)
Run RF to classify inflamed and control samples:
RF_state_classify <- randomForest( x=otu_table_scaled_state[,1:(ncol(otu_table_scaled_state)-1)] , y=otu_table_scaled_state[ , ncol(otu_table_scaled_state)] , ntree=501, importance=TRUE, proximities=TRUE )
Run RF to regress OTUs against inflammation score (IS):
RF_IS_regress <- randomForest( x=otu_table_scaled_IS[,1:(ncol(otu_table_scaled_IS)-1)] , y=otu_table_scaled_IS[ , ncol(otu_table_scaled_IS)] , ntree=501, importance=TRUE, proximities=TRUE )
Remember that if you partition your data into a training and test set beforehand (which we didn't do in this case due to low sample sizes) you would be able to directly validate your trained model on independent data. Without this partitioning the simplest way to assess model fit is to take a look at the model summaries by just typing the model name. For example, for the classification model:
RF_state_classify
Which returns:
Call: randomForest(x = otu_table_scaled_state[, 1:(ncol(otu_table_scaled_state) - 1)], y = otu_table_scaled_state[, ncol(otu_table_scaled_state)], ntree =501, importance = TRUE, proximities = TRUE)
Type of random forest: classification
Number of trees: 501
No. of variables tried at each split: 25
OOB estimate of error rate: 0%
Confusion matrix:
control inflamed class.error
control 20 0 0
inflamed 0 20 0
The out-of-bag error rate is 0%, which is perfect! Note that this summary also gives us the mtry parameter, which was 25 in this case. We can also take a look at the regression RF summary, which returns this (excluding the "Call" line):
Type of random forest: regression
Number of trees: 501
No. of variables tried at each split: 215
Mean of squared residuals: 0.02508508
% Var explained: 71.09
Note the much higher mtry parameter and that instead of out-of-bag error and a confusion matrix, we can use the mean of squared residuals and the % variance explained as performance metrics. These metrics suggest that this model is also performing extremely well although it isn't quite as clear as for the classification model.
Although the above performance metrics provided by the models were encouraging, often it isn't so clear that a model is working well. Testing whether your model's performance metric is more extreme than expected by chance based on a permutation test is one method to assess model significance.
To run these significance tests with 1000 permutations:
RF_state_classify_sig <- rf.significance( x=RF_state_classify , xdata=otu_table_scaled_state[,1:(ncol(otu_table_scaled_state)-1)] , nperm=1000 , ntree=501 )
RF_IS_regress_sig <- rf.significance( x=RF_IS_regress , xdata=otu_table_scaled_IS[,1:(ncol(otu_table_scaled_IS)-1)] , nperm=1000 , ntree=501 )
Both models are highly significant based upon these tests.
One method to estimate model performance is to systematically partition the data into training and test sets and repeatedly see how the model performs (cross-validation). There are many different ways to partition the data, but the simplest is leave-one-out cross-validation. The model is trained n times, where n is the number of samples, and one sample is left out each time for testing. This provides estimates of model performance, which tend to be quite similar to the internal measures (out-of-bag error and mean of squared residuals) described above. We will run this cross-validation using the caret R package.
The first step is defining the parameters we want to use for training:
fit_control <- trainControl( method = "LOOCV" )
In this case we are just specifying leave-one-out cross-validation, but caret can be used for much more sophisticated purposes.
These commands would run the leave-one-out cross-validation, note the same ntree and mtry parameters are being used as above:
RF_state_classify_loocv <- train( otu_table_scaled_state[,1:(ncol(otu_table_scaled_state)-1)] , y=otu_table_scaled_state[, ncol(otu_table_scaled_state)] , method="rf", ntree=501 , tuneGrid=data.frame( mtry=25 ) , trControl=fit_control )
RF_IS_regress_loocv <- train( otu_table_scaled_IS[,1:(ncol(otu_table_scaled_IS)-1)] , y=otu_table_scaled_IS[, ncol(otu_table_scaled_IS)] , method="rf", ntree=501 , tuneGrid=data.frame( mtry=215 ) , trControl=fit_control )
To see the performance metrics:
RF_state_classify_loocv$results
mtry Accuracy Kappa
1 25 1 1
RF_IS_regress_loocv$results
mtry RMSE Rsquared
1 215 0.1585135 0.7128165
Both the Accuracy and Kappa metrics are 100%, which should make us extremely confident in our classification model (you won't see models perform this well usually!). Also, the R-squared of the regression model is very similar to what we previously found based on the internal testing, which is typical since the internal RF performance metrics should not be biased.
par(mfrow=c(1,2))
RF_state_classify_imp <- as.data.frame( RF_state_classify$importance )
RF_state_classify_imp$features <- rownames( RF_state_classify_imp )
RF_state_classify_imp_sorted <- arrange( RF_state_classify_imp , desc(MeanDecreaseAccuracy) )
barplot(RF_state_classify_imp_sorted$MeanDecreaseAccuracy, ylab="Mean Decrease in Accuracy (Variable Importance)", main="RF Classification Variable Importance Distribution")
RF_IS_regress_imp <- as.data.frame( RF_IS_regress$importance )
RF_IS_regress_imp$features <- rownames( RF_IS_regress_imp )
RF_IS_regress_imp_sorted <- arrange( RF_IS_regress_imp , desc(`%IncMSE`) )
barplot(RF_IS_regress_imp_sorted$`%IncMSE`, ylab="% Increase in Mean Squared Error (Variable Importance)", main="RF Regression Variable Importance Distribution")
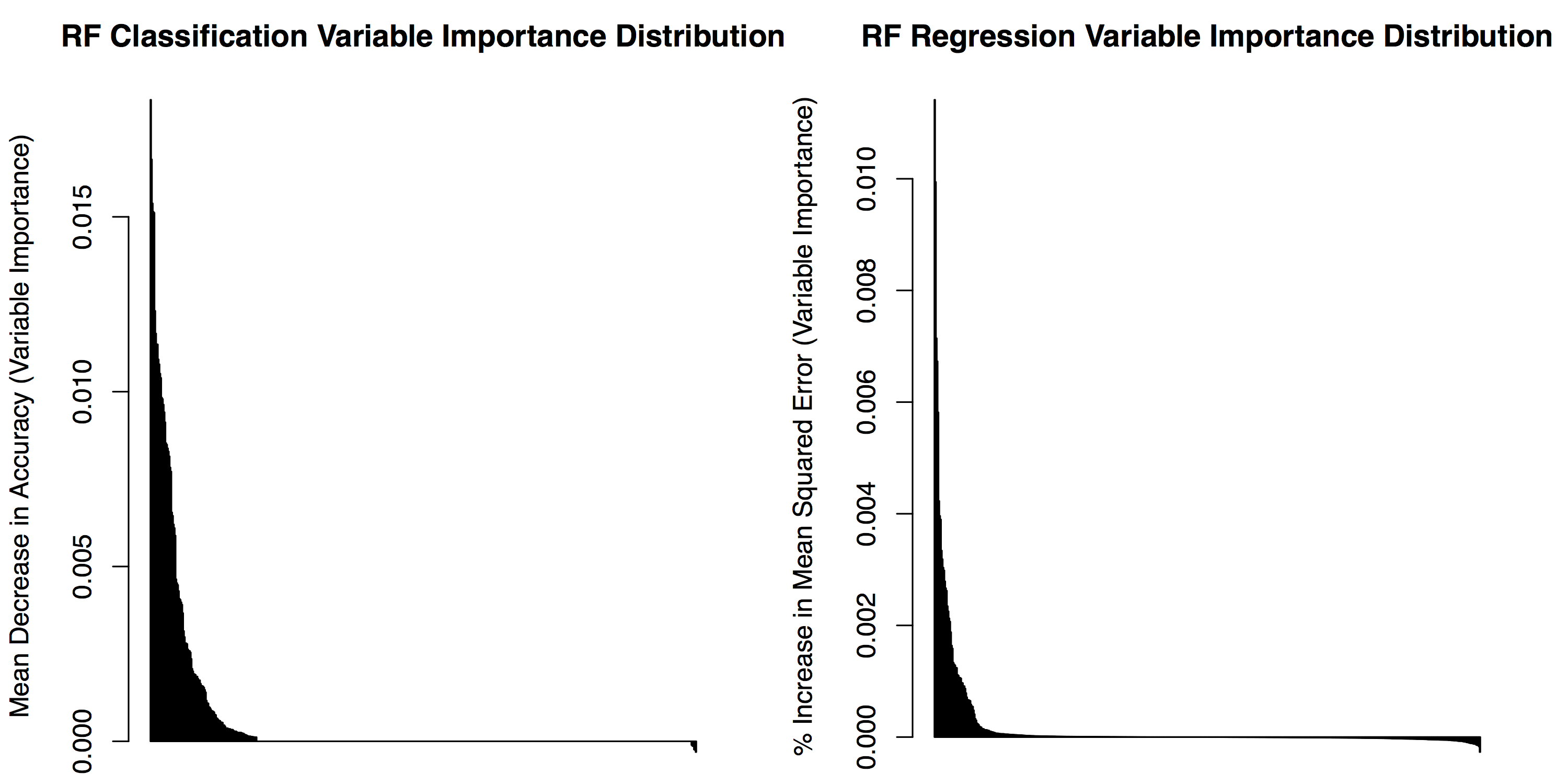
Often it's more informative to look at a subset of top features, for instance the top 10 features:
barplot(RF_state_classify_imp_sorted[1:10,"MeanDecreaseAccuracy"], names.arg=RF_state_classify_imp_sorted[1:10,"features"] , ylab="Mean Decrease in Accuracy (Variable Importance)", las=2, ylim=c(0,0.02), main="Classification RF")
barplot(RF_IS_regress_imp_sorted[1:10,"%IncMSE"], names.arg=RF_IS_regress_imp_sorted[1:10,"features"] , ylab="% Increase in Mean Squared Error (Variable Importance)", las=2, ylim=c(0,0.012), main="Regression RF")

Often it's better to save your R environment or particular R objects so that you don't need to re-run analyses. There are a few ways to do this, but what I find easiest is to use the saveRDS and readRDS functions. However, these functions restrict you to saving/reading one object at time. If you want to save many or all of the objects in your environment then you can just use the save and load functions. The issue with the save and load functions is that it can be easy to overwrite existing objects in your global environment, which is why I recommend the saveRDS and readRDS functions.
For instance, these commands will save each RF model objects to a .rda file:
saveRDS( file = "RF_state_model.rda" , RF_state_classify )
saveRDS( file = "RF_IS_model.rda" , RF_IS_regress )
Now if you create a new RStudio environment and go back to the same working directory as before you'll be able to re-load these objects (I re-named the objects to demonstrate that you don't need to use their previous names):
setwd("/Path/to/my/RF_tutorial/")
RF_state_model <- readRDS("RF_state_model.rda")
RF_IS_model <- readRDS("RF_IS_model.rda")