Microbiome Amplicon Sequencing Workflow
The following detailed protocol is for the generation of paired-end sequencing reads of 16S or 18S PCR amplicons with multiple barcodes (i.e.: “indices”) on the Illumina MiSeq machine of length ≈450 bp (300+300 bp with ~150 bp overlap) using v3 chemistry. It assumes an input of 380 individual samples (+4 controls = 384) in four 96-well plates (384-well plates are discouraged). Catalog numbers are given when items from specific vendors (vs. generic choices) are required.
Note that this workflow is continually being updated. If you want a specific version for printing (including the current one), select one of the PDFs below:
Link to Version 1 PDF (as originally published) - coming soon!
Last updated: 07 May 2020 (see "revisions" above for earlier versions)
- Single-channel pipettes (2-1000 µL)
- Multi-channel (8) pipette(s) (MCP; 2-100 µL)
- Required equipment for the DNA extraction kit(s) of your choice
- PCR machine with 96-well block
- Invitrogen Mother E-Base (#EB-M03) for E-Gels 96
- Documentation system for DNA gels (SYBR Safe filter)
- Microvolume DNA fluorescence reader (such as Invitrogen Qubit)
- Illumina Experiment Manager (iEM) software
- Illumina MiSeq sequencer with RTA v1.17.28 / MCS v2.2 or later
- Bead-mill / TissueLyser / Homogenizer (if using bead-based DNA extractions)
- Microvolume DNA spectrophotometer (such as NanoDrop; for quantifying extracts)
- Centrifuge with rotor for 96-well plates (useful for spinning down condensation)
- Standard gel electrophoresis system (for analyzing recalcitrant samples)
- Pipette filter tips (p2 to p1000)
- Reagent reservoirs for MCP
- 1.5 mL Eppendorf tubes
- DNA extraction kit(s)
- 96-well thin-wall 0.2 mL PCR plates (such as Bio-Rad #HSP9601)
- PCR plate films (such as Bio-Rad #MSB1001)
- Thermo Phusion High-Fidelity DNA Polymerase (#F-530L) or similar
- dNTP mix (at 40 mM = 10 mM of each base)
- Illumina fusion primers (see below and Excel template)
- PCR-grade water
- Invitrogen E-Gels 96 2% with SYBR Safe (#G7208-02)
- Invitrogen E-Gel Low Range Ladder (#12373-031; diluted 1:1 with PCR-grade water)
- Invitrogen SequalPrep Normalization Kit (#A10510-01) - cheaper alternative: Charm Just-a-Plate
- Invitrogen Qubit dsDNA HS reagent and assay tubes (clear 0.2 mL)
- 2 N NaOH
- 200 mM Tris-HCl, pH 7
- Illumina PhiX Control Kit v3 (#FC-110-3001)
- Illumina MiSeq Reagent Kit v3 (600 cycle) (#MS-102-3003)
- Ethanol (usually a requirement for extraction/purification kits)
- Mammalian blocking primer (see H. below; if substantial host contamination in 18S)
- Thin-wall 0.2 mL PCR tubes or strips with caps (for re-amplifying recalcitrant samples)
- Agarose, loading buffer/stain and 100 bp ladder (for analyzing recalcitrant samples)
- PCR product purification kit (if need to concentrate final library)
A. Use our Excel template to copy existing 16S/18S/ITS primers or to design your own custom gene primers with the proper Illumina indices and Nextera adapter orientations (download here). We order IDT “Ultramers” (www.idtdna.com) for such long primers (~80-90 nt) as their coupling efficiency is one of the highest available (critical for obtaining high proportions of full-length oligos in the mix you obtain). Order the fusion primers at 4 nmole scale in deep-well plates; one set per 96-well plate, arranged as follows, leaving blank rows in between sets:
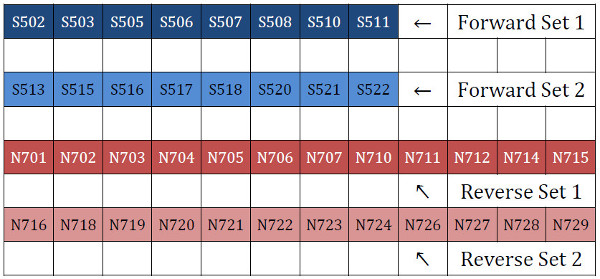
B. Once arrived, add 400 µL of PCR-grade water to each well containing the primers in order to reconstitute them at a concentration of 10 µM (1/10th the typical 100 µM working stock concentration for primers). We have found that these usually need a significant incubation time for the lyophilized pellets to re-suspend – we typically leave them overnight at 4°C.
C. Prepare the 1 µM working stock Forward Set 1 Primer Plate by pipetting 63 µL of PCR-grade water into each well of the 96-well PCR plate from a sterile reservoir. Rotate the deep-well primer plate 90° clockwise and align it so that the 8 occupied wells (= 8 different indices) of row 1 line up with the 8 rows of the new plate. Working by column and keeping the same set of tips, transfer 7 µL of reconstituted primer into each well of each column, mixing well by pipetting. Once complete, each column of the resulting plate will have enough primer for one complete 96-well plate PCR (leaving some extra for pipetting error; 12 columns × 5 µL = 60 µL required). Seal the plate with PCR film and store at -20°C.
D. Prepare the 1 µM working stock Forward Set 2 Primer Plate by repeating step C, but using row 3 of the reconstituted deep-well primer plate.
E. Prepare the 1 µM working stock Reverse Set 1 Primer Plate by pipetting 45 µL of PCR-grade water into each well of the 96-well PCR plate from a sterile reservoir. Align the deep-well primer plate horizontally (normal orientation) so that the 12 occupied wells (= 12 different indices) of row 5 line up with the 12 columns of the new plate. Working by row and keeping the same set of tips, transfer 5 µL of reconstituted primer into each well of each row, mixing well by pipetting. Once complete, each row of the resulting plate will have enough primer for one complete 96-well plate PCR (leaving some extra; 8 rows × 5 µL = 40 µL required). Seal the plate with PCR film and store at -20°C.
F. Prepare the 1 µM working stock Reverse Set 2 Primer Plate by repeating step E, but using row 7 of the reconstituted deep-well primer plate.
G. Once all aliquoting is complete, seal the deep-well plate with PCR film and archive at -20°C until new aliquots are required (minimized freeze-thaw cycles).
H. Optional: For the generation of 18S V4 amplicons from microbiome samples containing substantial non-target host DNA (ex: human, mouse, etc.), order a custom PNA mammalian blocking primer (elongation arrest in the V4 region courtesy of Laura Parfrey and Matt Lemay [UBC]) with the sequence: 5’-TCTTAATCATGGCCTCAGTT-3’. Once arrived, prepare an archival stock of 100 µM and a working stock of 10 µM using PCR-grade water.
-
Extract DNAs from your samples using the method/kit appropriate to the specific samples (ex: generally stool [with bead-beating kits], but also urine, etc.). For example, we have had good success with the MO-BIO PowerFecal Kit for mouse pellets and human stool. Note: Do not overload the kit columns with excessive amounts of sample material as this reduces extraction efficiencies and can allow contaminants/inhibitors to co-purify with the DNAs.
-
Quantify and quality-check your final DNAs via NanoDrop or Qubit to verify success. The A280/260 ratios should be 1.8 or better and our experience with community DNAs has shown that a concentration of at least 1 ng/µL is required to get consistent PCR results. Avoid costly secondary extractions/clean-ups for inhibitors (unless obvious) until PCRs below have truly shown inhibition – many sub-optimal DNAs will still perform adequately in PCR. Optional: A gel can be run to verify integrity (generally unnecessary for PCR-only studies).
-
Aliquot 5 µL of each of the 380 DNA samples into 4 × 96-well PCR plates (DNA Plates 1-4) in the order desired (we prefer by column), allowing for one PCR negative control well (position H12) on each plate; the layout of the primers (for ex: showing F1+R2 sets are used for Plate 2) are also indicated on this figure:

-
Prepare the following PCR master-mix for Plate 1 in a 1.5 mL Eppendorf tube (adjust if not using Phusion):

-
Dispense 78.5 µL of the master-mix into the 16 wells of 2 columns (or 105 µL in 12 wells of one row) of a 96-well plate (remaining wells to be used in subsequent PCR preps) – this plate now becomes the Master-Mix Plate and is used to transfer the master-mix into the PCR plate using an MCP.
-
Dispense 13 µL of master-mix into each well of the PCR Plate 1 (2 µL), one column (or row) at a time with the MCP.
-
Remove the protective film (“uncover”) from one column of the Forward Set 1 Primer Plate, align it horizontally on the bench to the left of PCR Plate 1 (2 µL) and dispense 5 µL into each well, one column at a time using the MCP. Note: You can use the same set of 8 tips for all.
-
Uncover one row of the Reverse Set 1 Primer Plate, align it vertically on the bench along the top of PCR Plate 1 (2 µL) and dispense 5 µL into each well, one row at a time using the MCP. Note: You must now change tips after every row to avoid cross-contamination (since different F primers/indices are now in each row).
-
Uncover the DNA Plate 1, align it along the top of PCR Plate 1 (2 µL) and dispense 2 µL into each well, one column at a time using the MCP. Note: Remember to change tips after every column.
-
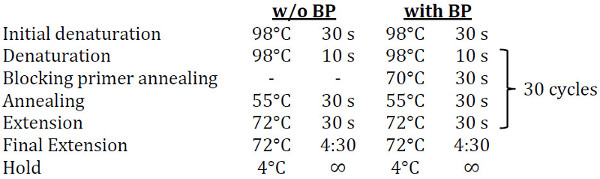
Once complete, seal the plate with PCR film, place in a thermocycler and run the following program (~1.5 h; adjust if not using Phusion):
-
While the first PCR is running, prepare the 1/10th dilution of DNA Plate 1 – add 27 µL of PCR-grade water to each well of the remaining 3 µL of template in DNA Plate 1, for a total of 30 µL final volume per well, using a reservoir (~3 mL required) and MCP. Note: Remember to change tips after every column.
-
Once the first PCR is complete (or nearly so), repeat steps 1-7 to prepare PCR Plate 1 (0.2 µL) using the newly diluted templates. Optional: Here, and for the subsequent plates below, once the two PCRs of the same plate are complete you can proceed to steps 3.1-3.4 and verify the PCR products before continuing with the next plate each time.
-
Once the two PCRs for Plate 1 are complete, repeat steps 1-9 to prepare PCR Plates 2 (2 µL) & (0.2 µL) from DNA Plate 2 using Forward Set 1 Primer Plate and Reverse Set 2 Primer Plate (note change to F1+R2 here).
-
Once the two PCRs for Plate 2 are complete, repeat steps 1-9 to prepare PCR Plates 3 (2 µL) & (0.2 µL) from DNA Plate 3 using Forward Set 2 Primer Plate and Reverse Set 1 Primer Plate (note change to F2+R1 here).
-
Once the two PCRs for Plate 3 are complete, repeat steps 1-9 to prepare PCR Plates 4 (2 µL) & (0.2 µL) from DNA Plate 4 using Forward Set 2 Primer Plate and Reverse Set 2 Primer Plate (note change to F2+R2 here).
-
Plug in the Mother E-Base, unwrap a fresh E-Gel 96 and insert it into the base.
-
The duplicate PCR reactions of Plate 1 are aggregated then loaded onto the gel in the same action: using the MCP and working by rows (the gel cannot be loaded by columns as they are staggered), pipette 20 µL out of the PCR Plate 1 (0.2 µL) into the corresponding wells of PCR Plate 1 (2 µL) and mix by pipetting up and down, then take 20 µL of this aggregate and load into the appropriate wells of the gel. Note: Remember to change tips after every row. Discard the empty PCR Plate 1 (0.2 µL) when finished and relabel the PCR Plate 1 (2 µL) the Aggregated PCR Plate 1.
-
Once all rows are complete, load 20 µL of the E-Gel Low Range Ladder into some of the marker (“M”) wells, then run the gel for the pre-set 12 min.
-
Visualize the gel and photograph on a UV/blue transilluminator with a SYBR filter. Example gels of 16S and 18S amplicons from this protocol are included in the PDF version of this document for reference.
-
Repeat steps 1-4 for PCR Plates 2 (2 µL) & (0.2 µL).
-
Repeat steps 1-4 for PCR Plates 3 (2 µL) & (0.2 µL).
-
Repeat steps 1-4 for PCR Plates 4 (2 µL) & (0.2 µL).
-
Any samples with failed PCRs (or spurious bands) are re-amplified by optimizing the PCR (further template dilution to 1:100 or using BSA/other additives) to produce correct bands in order to complete the amplicon plate. Unless this represents the majority of a plate (in which case continue with plates and E-gels), PCRs are done in standard tubes/strips and visualized using a traditional gel box. Once correct bands have been obtained, amalgamate those few tubes into the appropriate wells of the respective Aggregated PCR Plates before continuing.
-
Use the remaining 20 µL of each well in the Aggregated PCR Plate 1 to cleaned-up and normalize the amplicons using the high-throughput Invitrogen SequalPrep 96-well Plate Kit. Label this final plate SequalPrep Plate 1. Note: We have found it more convenient to inverse steps 1 & 2 of the SequalPrep protocol (i.e.: buffer first with one set of tips, then amplicons with another set). Only one set of tips (per step) is need in this kit as carry-over is minimal, amplicons are barcoded (therefore can’t “contaminate” each other’s reads) and amplicons will shortly be pooled anyways.
-
Once the SequalPrep protocol is complete, pool the 95 samples from SequalPrep Plate 1 by using the MCP to transfer 5 µL of each column into one column of a new 96-well plate named the Library Pool Plate (remaining columns to be used in subsequent pooling). Note: You can use the same set of 8 tips for all. Once complete, pipette 50 µL of each of the 8 wells into one 1.5 mL Eppendorf tube and label Plate 1 Library Pool.
-
Repeat steps 1-2 for Aggregated PCR Plate 2.
-
Repeat steps 1-2 for Aggregated PCR Plate 3.
-
Repeat steps 1-2 for Aggregated PCR Plate 4.
-
Once all four pools are complete, pipette 100 µL of each of the four tubes into one 1.5 mL Eppendorf tube and label Final Library Pool (add the run name to the tube or some other identifier to keep your various pools separate).
-
Quantify the Final Library Pool using the Invitrogen Qubit dsDNA HS assay (or similar fluorescence-based alternative; 5 µL of pool to be assayed) and calculate the molar concentration using the following formula, knowing that 1 ng/µL of a 500 bp amplicon = 3.29 nM:
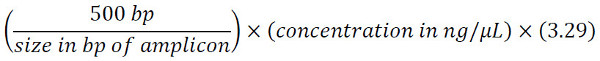
For the 16S amplicon generated using our V6-V8 primers (= 574 bp, including target region + adapters + indices) at a concentration of 3.0 ng/µL, for example: (500 bp / 574 bp) x 3.0 x 3.29 = 8.6 nM.
Note: We have found that the anticipated 1-2 ng/µL output from the SequalPrep plate is near impossible to achieve; we typically see concentrations in the range of 0.3-0.9 ng/µL.
-
This section is based upon the following Illumina documents, with some small procedural changes (including using the NextSeq variant for sample denaturation), and the inclusion of instructions to be able to load >96 samples (i.e.: 384 combinations of indices) which are not written out by Illumina – familiarize yourself with these documents / have them on-hand:
- MiSeq Reagent Kit v3 – Reagent Preparation Guide
- Preparing Libraries for Sequencing on the MiSeq
- Denaturing and Diluting Libraries for the NextSeq 500
- MiSeq System User Guide
-
Begin thawing the v3 Reagent Cartridge and tube of HT1 as instructed. Put at 4°C when complete. Optional: Take out the day before and thaw the reagents overnight at 4°C.
-
While waiting, prepare the Sample Plate and Sample Sheet files that will be used to run the MiSeq by opening the Illumina Experiment Manager (iEM) software.
-
Create the Sample Plates first – in order to run all 384 combinations of indices, 4 separate Sample Plates (one per plate from our protocol above) will be required. For the samples that were in DNA Plate 1:
- Choose Nextera XT v2 (Set A) in the iEM wizard.
- Give the plate a unique name (we usually use our run number and append an “A” to the end; ex.: 15A).
- Copy-and-paste the 96 sample names from your Excel sheet (after having brought the file over to the MiSeq via USB, Dropbox or email) into the Plate tab, then press Apply Default Index Layout. You will sometimes not see the index names show up on the top row and left-hand column of this tab, but if you switch to the Plate Graphic or Table views, they will be there correctly.
- Click on Finish and save the *A.nexxt28.plt file in the directory of your choice.
-
For the samples that were in DNA Plate 2, repeat step 4 except change to Nextera XT v2 (Set B) and append a “B” to the filename.
-
For the samples that were in DNA Plate 3, repeat step 4 except change to Nextera XT v2 (Set C) and append a “C” to the filename.
-
For the samples that were in DNA Plate 4, repeat step 4 except change to Nextera XT v2 (Set D) and append a “D” to the filename.
-
Now create the Sample Sheet by bringing in the 4 Sample Plates that belong to it:
- Select MiSeq in the iEM wizard, then Other --> FASTQ Only.
- Input your Reagent Cartridge barcode, select Nextera XT v2 for the Sample Prep Kit, input your Experiment Name (we usually use our complete run name now; ex.: IMR-Run15) and change the cycles to 301 for both reads.
- On the next screen, uncheck the Maximize box, choose Select Plate at left and navigate to and select your *A.nexxt28.plt file (Plate A).
- Once the samples are displayed, choose Select All + Add Selected Samples.
- Repeat the above two steps for the remaining plate files (Plates B/C/D).
- The Sample Sheet Status should show as Valid and, if so, click Finish to save the file, appending the run name to the end of the filename (for the above ex.: MSxxxxxxx-600V3-Run15.csv). We find it helpful having the run name/# when returning to the files, otherwise they are only labeled with the less-informative cartridge barcode by default. If your Sample Sheet Status is showing as Invalid, then it is often due to identical sample names being used in error. Once corrected, the status will update.
- It is a good idea to then simply verify that the CSV file is all correct by opening the file in WordPad and checking that the header information is correct (date, run name, FASTQ generation, etc.) and that you see the four sets of samples below in the [Data] section (Plate A samples, followed by Plate B, etc.).
-
Prepare 0.2 N NaOH as instructed, except make 10-fold less (20 µL of 1 N NaOH + 80 µL of water).
-
Use the nanomolar concentration from step 4.7 to determine whether the final amplicon library must be diluted or concentrated prior to continuing. A fixed concentration of 4 nM is the standard requirement for the MiSeq, however, using the NextSeq loading protocol, a library between 0.4-4 nM can be accommodated by simply using a larger volume of a more dilute library. Use the following guidelines:

-
Denature the library, using the indicated volume of 0.2 N NaOH in the table above, for 5 min. at room temperature.
-
Neutralize the reaction by adding the equivalent volume of Tris-HCl as indicated.
-
Dilute out the library to 20 pM using the indicated amount of chilled HT1 and place on ice.
-
Combine 570 µL of the library with 30 µL (=5%) of the already diluted and denatured PhiX Control Library. We always use 5% with amplicon libraries, regardless of anticipated diversity.
-
Proceed with loading the 600 µL sample in the v3 Reagent Cartridge and continue the MiSeq run start procedure, as instructed – the only slight change is that the default filename (just the cartridge barcode) of the Sample Sheet will not be found since we appended the run name to the end of the CSV file (for the above ex.: MSxxxxxxx-600V3-Run15.csv); browse to and select the correct file.