Metagenomics (taxonomic annotation; IMPACTT 2022)
This tutorial is for the 2022 IMPACTT bioinformatics workshop running from May 30th - June 1st. It has later been adapted to work for others that don't have access to the AWS server instance that was used for the workshop.
Authors: Robyn Wright and Morgan Langille
This tutorial has been adapted from the previous 2021 version written for the 2021 Canadian Bioinformatics Workshop by Jacob Nearing and Morgan Langille.
4. Generating Taxonomic Profiles with Kraken2 + Bracken
The main goal of this tutorial is to introduce students to different approaches for the taxonomic profiling of metagenomic data from microbiome samples. Throughout this tutorial we will emphasize that there is not a one-size-fits-all pipeline for analyzing MGS data. For example some individuals may opt to examine their data using a marker-based approach such as MetaPhlAn3 while others may opt to use a kmer based strategy such as Kraken2+Braken. Furthermore, in a subsequent module in this workshop students will also learn about another approach for examining microbiome data using metagenomic assembly and binning.
Throughout this tutorial there will be questions are aimed at helping students understand the various steps in reference based metagenomic profiling. The answers are found on this page.
This tutorial is based on our Metagenomics SOP and we are also working on a list of resources and papers that may be useful for beginners (or those that are not beginners!) in the microbiome field here.
- Become familiar with raw shotgun metagenomics data
- Learn how to use GNU Parallel to easily run commands on multiple files
- Learn common approaches to pre-processing MGS data
- Learn how to generate taxonomic profiles from your MGS data using both kmer based strategies Kraken2 + Bracken.
Properly citing bioinformatic tools is important to ensure that developers are getting the proper recognition. If you use any of the commands in this tutorial for your own work be sure to cite the relevant tools listed below!
- Bowtie2 (website, paper)
- FASTQC (website)
- GNU Parallel (website, paper)
- kneaddata (website)
- Microbiome Helper (website, paper)
- MinPath (website, paper)
- Kraken2 (website, paper)
- Bracken (website, paper)
- MMseqs2 (website, paper)
Anaconda, or for short conda, is a power tool used to managed different environments within your computer. Many bioinformatic tools can be installed using conda as it can significantly reduce the amount of leg work required when setting up the proper dependencies and versioning of tools. As such in this tutorial we have included a conda environment that contains all of the required bioinformatic tools!
You can activate this environment using the following command:
conda activate taxonomic
When you are finished this tutorial you can deactivate the conda environment using:
conda deactivate
If you are following this tutorial and are not in the IMPACTT workshop then you will need to setup the environments for yourself. You can do this by following these instructions for Linux (they will need slight modification if you are using a Windows computer, and I don't have a Windows computer to try them on - feel free to reach out if you have questions):
- Get and install Anaconda or Miniconda (this link was current at the time I made the tutorial, you can find the latest links here):
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
- Run the installation scripts:
bash Miniconda3-latest-Linux-x86_64.sh
Follow the prompts on the screen and then once it is installed, close and reopen your terminal. 3. Make the environments (I was using Python version 3.7.6 at the time, so I know that this works - you don't have to, but you may need to fix some errors if there are any incompatibilities in the package versions):
conda config --add channels bioconda
conda config --add channels conda-forge
conda create --name taxonomic python=3.7.6
conda create --name functional python=3.7.6
- Activate the taxonomic environment and install the packages that we will need:
conda activate taxonomic
conda install -c bioconda fastqc
conda install -c conda-forge parallel
conda install -c bioconda kneaddata
conda install -c bioconda kraken2
conda install -c bioconda bracken
conda deactivate
Follow any prompts that might come up on screen, usually you will just need to type y and press enter/return. 5. Activate the functional environment and install the packages that we will need:
conda activate functional
conda install -c bioconda mmseqs2
conda install numpy
conda install pandas
conda install ete3
conda deactivate
- Make and activate the Picrust2 environment (following the install from source instructions here:
wget https://github.com/picrust/picrust2/archive/v2.5.0.tar.gz
tar xvzf v2.5.0.tar.gz
cd picrust2-2.5.0/
conda env create -f picrust2-env.yaml
conda activate picrust2
pip install --editable .
pytest
cd ..
conda deactivate
- Activate the taxonomic environment again, that we will need for this part of the tutorial, and download the data that we will need:
conda activate taxonomic
wget -O metagenome_tutorial.tar.gz https://www.dropbox.com/sh/cn06suaq77di9sf/AADpERJGxme7xfON-rLoOE2Oa/metagenome_tutorial.tar.gz?dl=1
tar -xvf metagenome_tutorial.tar.gz
rm metagenome_tutorial.tar.gz
cd metagenome_tutorial
tar -xvf *.tar.gz
rm *.tar.gz
Often in bioinformatics research we need to run files corresponding to different samples through the same pipeline. You might be thinking that you can easily copy and paste commands and change the filenames by hand, but this is problematic for two reasons. Firstly, if you have a large sample size this approach will simply be unfeasible (in bioinformatics we are frequently working with hundreds or thousands of samples). Secondly, sooner or later you will make a typo! Fortunately, GNU Parallel provides a straight-forward way to loop over multiple files. This tool provides an enormous number of options: some of the key features will be highlighted below. We like using this handy cheatsheet with a range frequently used commands, or you can check out all of the options here.
The first step will be to create a directory that we will be working in. If you aren't in the IMPACTT workshop then you don't need to do this first part
cd workspace
mkdir Module4
cd Module4
Next we will need to download the zip folder containing our test data using the command wget:
wget https://www.dropbox.com/s/58grzx1ir7o8d3k/gnu_parallel_examples.zip?dl=1 -O gnu_parallel_examples.zip
unzip gnu_parallel_examples.zip
You can use wget to download the files that are at many links online, but if you copy a dropbox link like this one then you will need to change the =0 on the end to =1, like this one.
Note that the second command is just to unzip the .zip file.
You can explore the files that are in this folder using the ls command followed by the file path, for example:
ls gnu_parallel_examples/
ls gnu_parallel_examples/example_files/
ls gnu_parallel_examples/example_files/fastqs/
You should see that this has listed all of the files first in the gnu_parallel_examples/ and then in the gnu_parallel_examples/example_files/ folder and finally in the gnu_parallel_examples/example_files/fastqs/ folder.
We can now safely remove the zip file and move into the gnu_parallel_examples folder.
rm gnu_parallel_examples.zip
cd gnu_parallel_examples
Firstly, whenever you're using parallel it's a good idea to use the --dry-run option, which will print the commands to screen, but will not run them.
This command is a basic example of how to use parallel:
parallel --dry-run -j 2 'gzip {}' ::: example_files/fastqs/*fastq
This command will run gzip (compression) on the test files that are in example_files/fastqs/. There are 3 parts to this command:
- The options passed to GNU Parallel:
--dry-run -j 2. The-joption specifies how many jobs should be run at a time, which is 2 in this case. This means that both of the commands printed out will be run at the same time. This is what makes parallel so powerful as it allows you to run multiple commands on different files at the same time.
NOTE you should avoid trying to run more jobs at once than there are available processors on your workstation The workstations that we are working on have a total of 4 CPUs! As such we should not run parallel commands with more than 4 jobs at a time. This will vary depending on the hardware you are working on.
-
The commands to be run:
'gzip {}'. The syntax{}stands for the input filename that is the last part of the command. In parallel the command you want to run should always be surrounded by the character' -
The files to loop over:
::: example_files/fastqs/*fastq. Everything after:::is interpreted as the files to read in!
Remove --dry-run from the above command and try running it again. This should result in both fastq files located in example_files/fastqs/ to be compressed in gzip format. You can recognise this compression type as the files will always end in .gz.
If you list the files in the example_files/fastqs/ again using the ls command, you should see that now they are in gzip format.
Sometimes multiple files need to be given to the same command you're trying to run in parallel. As an example of how to do this, take a look at the paired-end FASTQ files in this example folder:
ls example_files/paired_fastqs
The "R1" and "R2" in the filenames specify forward and reverse reads respectively. If we wanted to concatenate the forward and reverse files together (paste them one after another into a single file for each pair) we could use the below command:
parallel --dry-run -j 2 --link 'cat {1} {2} > {1/.}_R2_cat.fastq' ::: example_files/paired_fastqs/*R1.fastq ::: example_files/paired_fastqs/*R2.fastq
There are a few additions to this command:
- Two different sets of input files are given, which are separated by
:::(the first set is files matching *R1.fastq and the second set is files matching *R2.fastq). - The
--linkoption will allow files from multiple input sets to be used in the same command in order.{1}and{2}stand for input files from the first and second sets, respectively. - Finally, the output file was specified as
{1/.}_R2_cat.fastq. The/will remove the full path to the file and.removes the extension.
It's important to check that each samples' files are being linked correctly when the commands are printed to screen with the --dry-run option. If the commands look OK then try running them without --dry-run now!
You should now have two files in your directory named test[1-2]_R1_R2_cat.fastq. Lets see how many lines these files contain with the following command:
wc -l test*
You can return to the higher directory with this command:
cd ..
Finally one thing to keep in mind when running commands with parallel is that you can keep track of their progress (e.g. how many are left to run and the average runtime) with the --eta option.
Alright now let's take a look at the data we'll be using for the main tutorial! The input files are FASTQ files, which contain information on the base-pairs in sequenced reads and the quality scores of each base-pair.
One thing we should be aware of when dealing with metagenomic data is that many tools will require a large amount of computational resources in order to run. As such they can either take a while to run or in some cases fail to run at all due to issues such as insufficient memory. In many cases it is unlikely that the metagenomic analysis of microbiome samples can be completed on a standard laptop in a timely manner.
For now we'll work with ten example sub-sampled FASTQs, that were downloaded from the HMP2 database that examined individuals with and without inflammatory bowel disease. These files are much smaller than normal metagenomic sequencing files but will speed up our data processing during this tutorial!
First we will need to copy the files from the storage section of our machine into our workspace. If you aren't in the IMPACTT workshop then you don't need to run this command
cp -r ~/CourseData/Module4/Data/raw_data/ .
Note the -r flag on the copy command indicates that we want to copy a directory and not an individual file and the . indicates that we are copying these into the current directory.
Take a look with less at the forward ("R1") and reverse ("R2") reads of one of the files.
less raw_data/CSM7KOMH_R1_subsampled.fastq.gz
less raw_data/CSM7KOMH_R2_subsampled.fastq.gz
You'll see that there are 4 lines for each individual read. Also, note that in the forward and reverse FASTQs for each sample that the read ids (the 1st line for each read) are the same and in the same order (except for /1 or /2 at the end).
To find out how many reads are in each forward FASTQ we can simply count the number of lines in a file and divide it by 4. Since the files are gzipped we'll need to use zcat to uncompress them on the fly and pipe (|) this as input to the wc -l command, which will return the number of lines in plaintext files.
zcat raw_data/CSM7KOMH_R1_subsampled.fastq.gz | wc -l
zcat raw_data/HSM6XRQY_R1_subsampled.fastq.gz | wc -l
We can see that the files have the same number of lines and therefore have the same read depths. However, this is not always the case with MGS data!
Question 1: Based on the above line counts can you tell how many reads should be in the reverse FASTQs? Remember that a standard convention is to mark the reverse FASTQs with the characters R2
Pre-processing is an important step when analyzing sequencing data. When not performed you could be allowing contaminants (such as DNA from the host) and/or low-quality reads to introduce noise to downstream analyses.
A standard first step with pre-processing sequence data is to visualize the base-qualities over the reads along with other helpful statistics. FASTQC is a simple program that will allow you to explore this information!
You can run FASTQC on the two test samples with this command (after making the output directory):
mkdir fastqc_out
fastqc -t 4 raw_data/*fastq.gz -o fastqc_out
This is a fairly simple tool and only has a few arguments.
-
-t- The number of threads we want to use to process each file -
raw_data/*fastq.gz- This indicates we want to run this command on all files that end withfastq.gzin the folder raw_data -
-o- Indicates the name of the folder we want to output the results to.
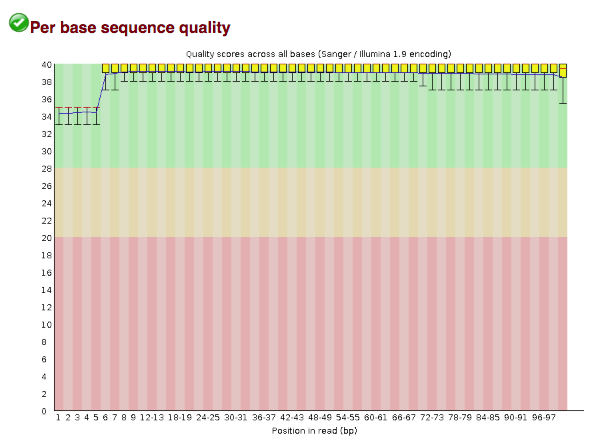
You can open one of the HTML files in your browser by entering the same address you used to connect to your AWS instance into your favourite web browsers and navigating to the workspace/Module4/fastqc_out/ folder. You should see something like this under the per-base quality section:
If you are not in the IMPACTT workshop then you will probably need to download this folder in order to view the files. The command should look something like this:
scp -r [email protected]:/home/robyn/metagenome_tutorial/fastqc_out/ .
These base qualities are not typical! Usually you would see the quality scores drop off near the end of the reads (and this tends to be even worse for reverse reads). However, it turns out that these reads have already been filtered! Importantly, this wouldn't have been obvious without visualizing the quality scores.
Even though these reads have already been through one round of quality filtering, we'll check them for contaminant sequences. Since these are human stool samples it's possible that there could be some human DNA that was sequenced (and could be high quality!). Also, sometimes PhiX sequences (a common sequencing control) aren't properly removed from output files. We'll want to remove both of these types of sequences before proceeding.
We'll use kneaddata, which is a wrapper script for multiple pre-processing tools, to check for contaminant sequences. Contaminant sequences are identified by this tool by mapping reads to the human and PhiX reference genomes with Bowtie2. You would also typically trim trailing low-quality bases from the reads at this step with Trimmomatic, which kneaddata will do automatically if you didn't use the skip-trim option below.
The below command will pass the read pairs to the same command, similar to how we ran them above. If you want to run quality processing as well then check out the command in our metagenomics SOP. Note that in this case we're running 1 job at a time (-j 1) and each job is being run over 4 threads (-t 4). This means we are using 4 processors (CPUs) at a time while running this program (the maximum number on your workstation!) The Bowtie2 index files have already been made and are passed to the tool with this option: -db ~/CourseData/Module4/Data/bowtie2_db/GRCh38_PhiX.
parallel --eta -j 1 --link 'kneaddata -i {1} -i {2} -o kneaddata_out/ \
-db /home/ubuntu/CourseData/Module4/Data/bowtie2db/GRCh38_PhiX \
-t 4 --bypass-trim ' \
::: raw_data/*_R1_subsampled.fastq.gz ::: raw_data/*_R2_subsampled.fastq.gz
If you are not in the IMPACTT workshop then your command should look like this:
parallel --eta -j 1 --link 'kneaddata -i {1} -i {2} -o kneaddata_out/ \
-db bowtie2db/GRCh38_PhiX -t 4 --bypass-trim ' \
::: raw_data/*_R1_subsampled.fastq.gz ::: raw_data/*_R2_subsampled.fastq.gz
If you take a peek in kneaddata_out with ls you'll see that a number of output files have been created! The key information we want to know is how many reads were removed. This command will generate a simple count table:
kneaddata_read_count_table --input kneaddata_out --output kneaddata_read_counts.txt
Question 2: Take a look at kneaddata_read_counts.txt: how many reads in sample CSM7KOMH were dropped?
You should have found that only a small number of reads were discarded at this step, which is not typical. It happens that the developers of IBDMDB have provided the pre-processed data to researchers so it would have been strange to find many low-quality reads.
Question 3: It's reasonable that the processed data was provided by IBDMDB since their goal is to provide standardized data that can be used for many projects. However, why is it important that normally all raw data be uploaded to public repositories?
Let's enter the kneaddata output folder now and see how large the output files are:
cd kneaddata_out
ls -lh *fastq
In this example case the output files are quite small (~5 megabytes), but for typical datasets these files can be extremely large so you may want to delete the intermediate FASTQs if you're running your own data through this pipeline. Also, note that the output FASTQs are not gzipped - you could gzip these files if you planned on saving them long-term. In the meantime you can move the intermediate FASTQs to their own directory with these commands:
mkdir intermediate_fastqs
mv *_contam*fastq intermediate_fastqs
mv *unmatched*fastq intermediate_fastqs
cd ..
If you want to look at all of the steps that were run by kneaddata then you can take a look at the .log files in the kneaddata_out folder using the less command.
We have one last step before we are ready to begin generating taxonomic profiles! At this point we have reached a fork in the road where we have two options. The first option is to stitch our forward and reverse reads together while the second is to simply concatenate (combine) the forward and reverse reads into a single sequencing file.
The process of stitching involves taking the forward read and combining it with the reverse complement of the reverse read based on where the reads would overlap in the middle.
This process can result in the majority of your reads being thrown out during the stitching process as many metagenomic reads will have little to no overlap. This is because in many cases during library preparation the DNA fragments are too large for the forward and reverse reads to completely traverse them. Remember that the forward and reverse reads only cover 100-150bp depending on platform and DNA fragments from many metagenomic library preps average in size between 300-500bp. This means there is not sufficient overlap to bring the forward and reverse reads together. As such I would suggest skipping this step to avoid losing a majority of reads.
If you would like to read more about how you might go about the stitching process using the program PEAR check out this page here.
Instead of dealing with the issue of stitching we will instead concatenate (combined) the forward and reverse reads into a single file. This is why its important during our filtering steps to only keep reads that both pairs passed all filtering parameters. If not we might end up with unaccounted for bias due to more forward reads being included in our concatenated reads than reverse.
We can do this by running the command: If you are not in the IMPACTT tutorial then you can skip the second line of this
conda deactivate
ln -s ~/CourseData/Module4/Data/helper_scripts/ .
perl helper_scripts/concat_paired_end.pl -p 4 --no_R_match -o cat_reads kneaddata_out/*_paired_*.fastq
conda activate taxonomic
Note the first command is simply linking the folder that contains the script for running this command to the current directory we are working in. We also had to deactivate and reactivate the environment that we were using - we often find in bioinformatics that some programs will need different versions of packages in order to work, so having them installed in different environments is one way to manage this.
Always double check the output to make sure the correct files are being concatenated together
Now that we have processed all of our reads the next step is to use Kraken2 to classify them with taxonomic labels. Kraken2 is one of many popular tools for classifying metagenomic reads into taxonomic profiles. In this case Kraken2 uses a kmer approach to assign reads to specific taxonomic lineages (look back at the lecture slides to remind yourself how kmers are used to classify reads).
There are several choices that we can make when classifying our samples that can have a very significant impact on both the proportion of reads in samples that we are able to classify as well as the taxa that are classified (if a taxon doesn't exist in our database, then we won't be able to classify it in our samples). The biggest choices here are the reference database that we use as well as the confidence threshold. We have recently published a preprint that addresses this in some detail, but we'll summarise some of the most important points below.
As it will take a few minutes for Kraken2 to run, we will just start running the commands and then there are a few things that we should understand about what we're doing.
We are going to use our knowledge about parallel from above to process both of our samples through kraken2 one after another!
We will first make directories to hold our results:
mkdir kraken2_outraw
mkdir kraken2_kreport
When you're running Kraken2 you should always be careful that you've created these folders before you run it! It won't give you an error message telling you that the folders don't exist, but it won't be able to create any output files so your time running it will have been wasted if you don't have the folders made.
Next we will run the Kraken2 command using parallel. Notice here that we now have two arguments going into parallel with ::: at the end, so they are named {1} and {2} inside the command, as we discussed above.
parallel --dry-run -j 1 --eta 'kraken2 --db ~/CourseData/Module4/Data/kraken_db/k2_standard_8gb_20210517/ --threads 4 --output kraken2_outraw/{1/.}_{2}_minikraken.kraken.txt --report kraken2_kreport/{1/.}_{2}_minikraken.kreport --use-names {1} --confidence {2}' ::: cat_reads/*.fastq ::: 0.0 0.1
If you are not in the IMPACTT workshop, run this command instead:
parallel --dry-run -j 1 --eta 'kraken2 --db k2_standard_8gb_20210517/ --threads 4 --output kraken2_outraw/{1/.}_{2}_minikraken.kraken.txt --report kraken2_kreport/{1/.}_{2}_minikraken.kreport --use-names {1} --confidence {2}' ::: cat_reads/*.fastq ::: 0.0 0.1
-
--db- This option points to the location of the database you want Kraken2 to use for classification. We have already set this AWS instance with the minikraken database. -
--threads 4-This option indicates that we want to use four processors for each Kraken2 job -
--output- This argument points to the file name we want to output our classifications to. -
--report- This argument points to the file name we want to output a more detailed classifcation report for each sample. These more in-depth classification reports are required to correct abundance estimates using bracken. -
--use-names- This indicates we want the classifier to output the taxon names rather than their NCBI IDs. -
--confidence- This allows us to set the confidence threshold so as reads with a low proportion of their k-mers classified as the taxon will be unclassified in the output (see below).
Question 4: What does the text {1/.}.kraken.txt and {1/.}.kreport do in the above command? (Hint look at our above discussion of parallel).
Check to make sure the command you are running is the one you expected using the --dry-run flag. Once you are sure remove the flag and run the command.
Some summary information will be output to the console when you run this command including the number of sequences that were classified versus unclassified in each sample.
Unlike metagenomic assembly, we will be classifying our sequenced reads based on how similar they are to reads we have already classified in a reference database. In the Kraken2 manual, the Kraken2 developers recommend building a "standard" database that is comprised of the bacterial, archaeal and viral domains as well as the human genome and a collection of known vectors (UniVec_Core). There are commands given in the manual for doing this, and a relatively up-to-date version of this database is also available for download via the Langmead lab here. This database requires approximately 100 Gb RAM/memory to build, and 40 Gb RAM to run. The Langmead Lab also provides some secondary databases that are capped at different sizes, and also that contain other domains in addition to the bacteria, archaea and viruses. In our preprint, we found that the best database to use with a range of simulated and mock samples (where the real composition of the sample was known) was one that contained all genomes from the NCBI Reference Sequence Database (NCBI RefSeq), and can be downloaded from here. However, this database is approximately 1.2 Tb and will therefore be too large for many researchers to use. We found that bigger databases weren't always better, and without access to a server with very large amounts of RAM, the best options were smaller, curated databases - we recommend that everyone thinks carefully about this choice prior to use on their own samples, and provided some guidance on this in the preprint.
Because we only have 16 Gb of RAM/memory on our instances here, we will be using the Standard-8 database (standard database capped at 8 Gb, which we'll call the minikraken database). The smaller size means that we can run it on workstations with a lower amount of RAM, but this comes at the cost of reduced performance. Also note that by default, kraken uses NCBI taxonomy (and requires a taxonomy structure similar to that of the NCBI taxonomy, where all taxa at all ranks are assigned a taxonomic ID, and the "parent" taxon is known for all), however, there are a number of other databases that users have made using different styles of taxonomy such as the Genome Taxonomy Database.
As mentioned above, Kraken2, uses exact alignment of k-mers (sub-sequences of length k) to a reference database for query read classification. Because k-mers within a read could map to multiple sequences within the database when using Kraken2, a lowest common ancestor (LCA) approach is used to classify the sequence, and the number of k-mers mapping to a given taxon are calculated as a proportion of the number of k-mers within that sequence that do not have ambiguous nucleotides. This proportion of k-mers mapping to a taxon is called the “confidence”, and there is a confidence threshold that can be set within the program (0 by default), where a taxonomic classification is only taken if it is above this pre-defined threshold. For a short example of this, let's look at the below figure (taken from the original Kraken paper):

Here, we can see that the query sequence has been broken up into 27 k-mers. 11 of these k-mers are unclassified (shown in white), while 10 have been classified as the blue taxon, 4 as the orange taxon, 1 as the purple taxon and 1 as the red taxon. In this example, the purple taxon is the ancestor of all taxa and the blue is the ancestor of the orange. The sequence will be classified to the leaf node, or the lowest rank possible, and we can use the number of k-mers that were classified to calculate how confident we are in that taxonomic assignment. Our confidence that this sequence belongs to the orange taxon if 4/27, or 0.15. Our confidence that it belongs to the blue taxon is (10+4)/27 = 14/27 = 0.52, and our confidence that it belongs to the purple taxon is the sum of all classified k-mers, so 16/27 = 0.59. By default, the confidence threshold that Kraken uses is 0.00, which would mean that the sequence would be classified as the orange taxon. If we increased the confidence threshold to 0.20 then the sequence would be classified as the blue taxon, and if we increased it to 0.60 or higher then the sequence would be unclassified.
Again, you can see the preprint if you want to see how this impacts the classifications given in more detail (and for more guidance on this), but as the confidence threshold is increased, fewer reads will be classified, but we can be more certain that the reads that are classified are classified correctly. To some extent, the "best" confidence threshold to use will depend on your research question and whether you are more concerned with ensuring that you identify all taxa that you possibly can (no false negatives), or that all of the taxa that you do identify are actually in the sample (no false positives). If you are running your own samples, then it may be worth taking a small (representative) subset and running them with a few different thresholds to see how the confidence threshold impacts this.
Hopefully now that you've read about databases and confidence thresholds, the Kraken2 commands have finished running.
Now we will take a look at some of the output files from Kraken2. First take a look at one of the files in the kraken2_kreport folder with the less command. The first few lines should look something like this:
31.03 14897 14897 U 0 unclassified
68.97 33117 14 R 1 root
68.94 33102 0 R1 131567 cellular organisms
68.94 33101 588 D 2 Bacteria
64.21 30828 4 D1 1783270 FCB group
64.20 30824 0 D2 68336 Bacteroidetes/Chlorobi group
64.20 30824 334 P 976 Bacteroidetes
63.50 30489 2 C 200643 Bacteroidia
63.50 30487 1519 O 171549 Bacteroidales
60.06 28835 0 F 815 Bacteroidaceae
60.06 28835 18479 G 816 Bacteroides
10.64 5108 5106 S 28116 Bacteroides ovatus
0.00 2 2 S1 1379690 Bacteroides ovatus V975
The columns here are (from the Kraken2 manual):
| Column Number | Data Type |
|---|---|
| 1 | The percentage of sequences covered by the clade rooted at this taxon |
| 2 | The number of sequences covered by the clade rooted at this taxon |
| 3 | The number of sequences assigned directly to this taxon |
| 4 | A rank code, indicating (U)nclassified, (R)oot, (D)omain, (K)ingdom, (P)hylum, (C)lass, (O)rder, (F)amily, (G)enus, or (S)pecies |
| 5 | NCBI taxonomic ID number |
| 6 | Indented scientific name |
In the example output above, 31.03% of sequences were unclassified while 68.97% of sequences were classified. The output is hierarchical, so going down, we can see that 68.94% of sequences were from cellular organisms (so almost all of the classified sequences), and 68.94% were Bacterial sequences. If we look at the second and third columns for the Bacteria, we can see that 588 of the 33,101 sequences that were classified as Bacteria weren't classified to any rank lower than Bacteria (Domain rank).
Because the full RefSeq database (RefSeqCompleteV205) is far too big to use on these instances, we've run this with these samples for you so that you can see the difference. Go ahead and copy them across to your directory:
cp ~/CourseData/Module4/Intermediate_Results/kraken2_kreport/*RefSeqCompleteV205.kreport kraken2_kreport/
cp ~/CourseData/Module4/Intermediate_Results/kraken2_outraw/*RefSeqCompleteV205* kraken2_outraw/
If you are not in the IMPACTT workshop then you can run this command instead:
cp kraken2_kreport_RefSeqCompleteV205/*RefSeqCompleteV205.kreport kraken2_kreport/
cp kraken2_outraw_RefSeqCompleteV205/*RefSeqCompleteV205* kraken2_outraw/
You can see that these samples have RefSeqCompleteV205 (which includes the version of the NCBI RefSeq database that we used to make the Kraken2 database) in the file names rather than minikraken.
Question 5: How many more reads in each sample are classified using the full RefSeq database (RefSeqCompleteV205) than with the minikraken database (with no confidence threshold)?
*Hint: you can use the less command to look at both the .kreport and the .kraken.txt files.
Question 6: How many more reads in each sample are classified using a confidence threshold of 0.0 compared with a confidence threshold of 0.1 for the minikraken database?
We will also take a quick look at the raw output files from Kraken2. Go ahead and look at the file kraken2_outraw/CSM7KOMH_0.0_minikraken.kraken.txt with the less command. The first few lines should look something like this:
C CB1APANXX170426:1:1101:10000:16732/1 Bacteroides (taxid 816) 101 0:17 816:4 0:29 816:4 0:13
U CB1APANXX170426:1:1101:10000:4761/1 unclassified (taxid 0) 101 0:67
C CB1APANXX170426:1:1101:10001:37312/1 Bacteroides (taxid 816) 101 0:57 816:5 0:4 816:1
U CB1APANXX170426:1:1101:10001:68734/1 unclassified (taxid 0) 50 0:16
U CB1APANXX170426:1:1101:10002:26975/1 unclassified (taxid 0) 101 0:40 2763022:1 0:26
U CB1APANXX170426:1:1101:10002:36688/1 unclassified (taxid 0) 68 0:34
This "raw" output has a line for each sequence, and the columns here are:
| Column Number | Data Type |
|---|---|
| 1 | C/U for Classified/Unclassified |
| 2 | The sequence name (taken from the FASTQ file) |
| 3 | The taxon that this sequence was classified as |
| 4 | The length (in base pairs) of the sequence |
| 5 | A list indicating the mapping of each minimizer in the sequence |
For column 5, in this example, the first 17 minimizers were unclassified (taxid 0), the next four minimizers were classified as Bacteroides (taxid 816), the next 29 minimizers were unclassified, the next four minimizers were classified as Bacteroides, and the last 13 minimizers were unclassified. In this case, almost all of the minimizers were unclassified while only a eight minimizers were classified, so this read would have a confidence of 8/(17+4+29+4+13) = 0.119 that it is Bacteroides.
Question 7: Take a look at the raw output for the same sample but run with a confidence threshold of 0.1. Was the first read classified? Why or why not?
The raw output files created by Kraken2 should be corrected using Bracken to get more accurate estimations on the abundance of different taxa at various taxonomic levels. While Kraken2 classifies each of our reads to their best locations within a taxonomic tree, in some cases this will result in a read being placed higher up in the tree due to two species or sometimes even genera sharing the exact same sequence (k-mer) to which that read matched. Bracken can be used to solve this issue by taking these reads and mapping them back to the genomes they might belong to. By doing this it generates new abundance estimates that take into account the problem laid out above.
To apply Bracken to our samples to estimate the species level abundances we can run the following commands:
First we will make a directory to output the results to:
mkdir bracken_out
Now we will run the bracken command:
parallel --dry-run -j 2 'bracken -d ~/CourseData/Module4/Data/kraken_db/k2_standard_8gb_20210517/ -i {} -o bracken_out/{/.}.species.bracken -r 100 -l S -t 1' ::: kraken2_kreport/*minikraken.kreport
If you are not in the IMPACTT workshop, run this command instead:
parallel --dry-run -j 2 'bracken -d k2_standard_8gb_20210517/ -i {} -o bracken_out/{/.}.species.bracken -r 100 -l S -t 1' ::: kraken2_kreport/*minikraken.kreport
-
-i-The input kreport file from kraken2 -
-r- The length of the reads used for mapping -
-l- The level at which to estimate taxon abundance (S indicates species) -
-t- The number of reads required for a taxon to be used in the abundance estimate
Note generally you would increase the -t argument to be somewhere between 0.1-1% of the total average read depth, however, since we are working with files that have already been subset we will not set this option. The reason for this is that k-mer based strategies such as kraken2 are notorious for making a large number of low abundance false positives. Check-out this paper or our preprint for more information.
The final step in generating our taxonomic profiles is collecting all the results from each sample into a single file. This can be done using a helper script that comes installed with bracken!
mkdir bracken_out_merged
python helper_scripts/combine_bracken_outputs.py --files bracken_out/*minikraken.species.bracken -o bracken_out_merged/merged_output_minikraken.species.bracken
Lets take a look at our resulting file with the less command. You will notice that the file has 7 different columns corresponding to different information about a single taxon.
| Column Name | Data Type |
|---|---|
| name | Taxonomic ID |
| taxonomy_id | NCBI taxonomic ID |
| taxonomy lvl | Character representing the level of taxonomy |
| SAMPLE.species.bracken_num | Number of reads that aligned to this classifcation for this SAMPLE |
| SAMPLE.species.bracken_frac | Proportion of reads that aligned to this classification for this SAMPLE |
| SAMPLE2.species.bracken_num | Number of reads that aligned to this classifcation for this SAMPLE2 |
| SAMPLE2.species.bracken_frac | Proportion of reads that aligned to this classification for this SAMPLE2 |
| etc. |
We can also copy across the bracken files for the RefSeqCompleteV205 database:
cp ~/CourseData/Module4/Intermediate_Results/bracken_out/*RefSeqCompleteV205* bracken_out/
cp ~/CourseData/Module4/Intermediate_Results/bracken_out_merged/*RefSeqCompleteV205* bracken_out_merged/
If you are not in the IMPACTT workshop, use this command instead:
cp bracken_out_RefSeqCompleteV205/*RefSeqCompleteV205* bracken_out/
cp bracken_out_merged_RefSeqCompleteV205/*RefSeqCompleteV205* bracken_out_merged/
Question 8: How would you go about figuring out the total number of species we found within our ten different samples?
Question 9: How does the number of species classified by each of the confidence thresholds differ, and how does it differ between the minikraken and the RefSeqCompleteV205 databases?