DADA2 16S Chemerin Tutorial
This tutorial outlines how to process 16S rRNA sequencing data with the DADA2 pipeline. This tutorial is based on the DADA2 Big Data Tutorial and our previous 16S tutorial. We have written R scripts to allow the DADA2 pipeline to be run from the command-line.
Author: Gavin Douglas
First created: Sept 2017
- Basic unix skills (This is a good introductory tutorial: http://korflab.ucdavis.edu/bootcamp.html).
- The exact commands we'll be running assume that you're running this tutorial on our Ubuntu Desktop virtual image. If you are running it elsewhere just be aware you will need to change the file paths. If you are not using our virtual box you will need:
Amplicon sequencing is a common method of identifying which taxa are present in a sample based on amplified marker genes. This approach contrasts with shotgun metagenomics where all the DNA in a sample is sequenced. The most common marker gene used for prokaryotes is the 16S ribosomal RNA gene. It features regions that are conserved among these organisms, as well as variable regions that allow distinction among organisms. These characteristics make this gene useful for analyzing microbial communities at reduced cost compared to shotgun metagenomics approaches. Only a subset of variable regions are generally sequenced for amplicon studies and you will see them referred to as V3-V4 (i.e. variable regions 3 to 4) in scientific papers. For this tutorial we will process V6-V8 amplified sequences.
This tutorial dataset was originally used in a project to determine whether knocking out the protein chemerin affects gut microbial composition. Originally 116 mouse samples acquired from two different facilities were used for this project (only a subset of samples were used in this tutorial dataset, for simplicity). Metadata associated with each sample is indicated in the mapping file (map.txt). In this mapping file the genotypes of interest can be seen: wildtype (WT), chemerin knockout (chemerin_KO) and chemerin receptor knockout (CMKLR1_KO). Also of importance are the two source facilities: "BZ" and "CJS". It is generally a good idea to include as much metadata as possible, since this data can easily be explored later on.
If you are running this tutorial as part of EMBL-EBI's Metagenomics workshop then the tutorial files should already be on your computer. You can copy the zipped folder to the Desktop, unzip it, and enter the folder to get started.
cd ~/Desktop
cp ~/Documents/16S_chemerin_tutorial.zip ~/Desktop
unzip 16S_chemerin_tutorial.zip
cd 16S_chemerin_tutorial
Otherwise you can download the tutorial files by replacing the cp command above with the below wget command.
wget http://kronos.pharmacology.dal.ca/public_files/tutorial_datasets/16S_chemerin_tutorial.zip
Before going any further we will need to specify the anaconda environment to work in, which is a helpful package manager. Using different anaconda environments is convenient on systems where there could be different versions of programs used for different projects. In this case we will be using the qiime1 environment, which is already set-up.
source /anaconda2/bin/activate qiime1
Note when you want to deactivate this environment you can type:
source /anaconda2/bin/deactivate
DADA2 infers the error rates from the reads themselves and will have trouble if primer sequences are included so we first need to remove them. We will do so by running cutadapt on each pair of FASTQs with GNU parallel.
Before running all FASTQs in parallel however, it may be helpful to look at a single cutadapt command, which you do not need to run. This command will first determine if the matching primer sequences arefound at the beginning of each read in a pair (with up to 10% of bases differing) and if so this pair will be trimmed and written to output.
Note: the \ characters in the below commands are just used to break up commands over multiple lines for clarity. These are not necessary to use for your own commands.
cutadapt \
--pair-filter any \
--no-indels \
--discard-untrimmed \
-g ACGCGHNRAACCTTACC \
-G ACGGGCRGTGWGTRCAA \
-o primer_trimmed_fastqs/109CHE6WT_S369_L001_R1_001.fastq.gz \
-p primer_trimmed_fastqs/109CHE6WT_S369_L001_R2_001.fastq.gz \
fastq/109CHE6WT_S369_L001_R1_001.fastq fastq/109CHE6WT_S369_L001_R2_001.fastq
This command is being run on paired FASTQs: 109CHE6WT_S369_L001_R1_001.fastq and 109CHE6WT_S369_L001_R2_001.fastq. The options mean:
-
--pair-filter any: if either read fails the filtering criterion, both will be discarded. -
--no-indels: no INDELs are permitted in primer sequences. -
--discard-untrimmed: discard reads that do not contain the matching sequence. -
-gand-G: the forward and reverse primer sequences to be matched respectively (note they contain IUPAC characters).
parallel is a handy way to get around writing out this command for each pair of reads. It can be slightly confusing at first, but it is extremely useful once you master it! You can see a brief tutorial on GNU parallel here.
The below command will run cutadapt on all FASTQs. The --link option is extremely important in this case as it specifies that there are two input files to each parallel command.
Running parallel with the --dry-run option is a good way to double-check that the commands are being specified correctly (they will be printed to screen, but not run).
mkdir primer_trimmed_fastqs
parallel --link --jobs 9 \
'cutadapt \
--pair-filter any \
--no-indels \
--discard-untrimmed \
-g ACGCGHNRAACCTTACC \
-G ACGGGCRGTGWGTRCAA \
-o primer_trimmed_fastqs/{1/}.gz \
-p primer_trimmed_fastqs/{2/}.gz \
{1} {2} \
> primer_trimmed_fastqs/{1/}_cutadapt_log.txt' \
::: fastq/*_R1_*.fastq ::: fastq/*_R2_*.fastq
Each sample's logfile created above can be parsed to put the counts of passing reads into a single table (cutadapt_log.txt by default).
parse_cutadapt_logs.py \
-i primer_trimmed_fastqs/*log.txt
We will now be filtering out low quality reads from the dataset. To get an idea of what cut-offs should be used you should always take a look at the quality distribution across your reads. The commands below will generate a quality report per sample which you can open in a web browser.
mkdir primer_trimmed_fastqc_out
fastqc -t 9 primer_trimmed_fastqs/*fastq.gz -o primer_trimmed_fastqc_out/
You should see a plot like this for the distribution of quality scores per base-pair in forward reads:

And like this for the reverse reads:

Notice that the reverse reads' quality scores dips off much sooner than the forward reads, which is typical for Illumina sequencing.
dada2_filter.R \
-f primer_trimmed_fastqs --truncLen 270,210 --maxN 0 \
--maxEE 3,7 --truncQ 2 --threads 9 --f_match _R1_.*fastq.gz \
--r_match _R2_.*fastq.gz
The output read count table (dada2_filter_read_counts.txt by default) is helpful to look at to help determine if cut-offs were too lenient or stringent.
The output filtered reads are written to filtered_fastqs/ by default.
Now that we have our filtered reads in hand, we are ready to actually infer the error models from the reads and identify "variants" (as opposed to "OTUs"). Paired-end reads will also be merged at this step.
dada2_inference.R \
-f filtered_fastqs/ --seed 4124 -t 9 --verbose --plot_errors
Options:
-
-f filtered_fastqs/: input reads. -
--seed 4124: random seed which should be set for reproducibility. -
--plot_errors: output plots of the inferred error rates (for sanity check).
The output R object containing the inferred variants and read counts per sample is written to seqtab.rds by default. This is a binary object that can be read into R by downstream steps or by yourself with the readRDS command in R.
Once again it is helpful to look at the read counts in the logfile (dada2_inferred_read_counts.txt by default). It is typical to see some drop in reads at the merging step, but if this is too restrictive then you may need to change the upstream filtering cut-offs.
The final DADA2 steps are to remove chimeric variants and assign taxonomy to the variants.
dada2_chimera_taxa.R -i seqtab.rds \
-r ~/Documents/dada2_rdp_ref/rdp_train_set_16.fa.gz \
--skip_species -t 9
The --skip_species option means that only taxonomy assignment until the genus level will be outputted (when this option is not set an additional output file is created).
The above command created a sequence count table written to seqtab_final.rds by default. The logfile dada2_nonchimera_counts.txt is again helpful to look at to see how many reads and variants were discarded at the chimera checking step.
The above commands each generated a logfile. We have written an Rscript that will take in logfiles in this format and combine them into one as shown by the below command.
merge_logfiles.R \
-i cutadapt_log.txt,dada2_filter_read_counts.txt,dada2_inferred_read_counts.txt,dada2_nonchimera_counts.txt \
-n cutadapt,filter,infer,chimera -o combined_log.txt
At this point there are a number of different analysis workflows you could use. The DADA2 authors recommend phyloseq, which you can read about in their tutorial. Here, we will present a more straight-forward workflow using QIIME.
To begin we first need to convert the sequence count table into a BIOM and FASTA file respectively. The --taxa_in option also specified taxonomic identifiers which will be outputted per DADA2 variant into a textfile.
convert_dada2_out.R -i \
seqtab_final.rds -b seqtab.biom -f seqtab.fasta --taxa_in tax_final.rds
The taxonomy labels can be added as metadata to the BIOM file using the below command.
biom add-metadata -i seqtab.biom -o seqtab_tax.biom --observation-metadata-fp \
taxa_metadata.txt --observation-header OTUID,taxonomy --sc-separated taxonomy
Now that we have our BIOM file we can see a summary report with the below command. This is especially useful for choosing a reasonable rarefaction cut-off in the next step.
biom summarize-table -i seqtab.biom -o seqtab_summary.txt
Based on the above report most samples have > 200 reads so we will rarify to this depth. Note that this is extremely low depth and in most cases you should not be using a cut-off this low on your own data.
single_rarefaction.py -i seqtab_tax.biom -o seqtab_tax_rarified.biom -d 200
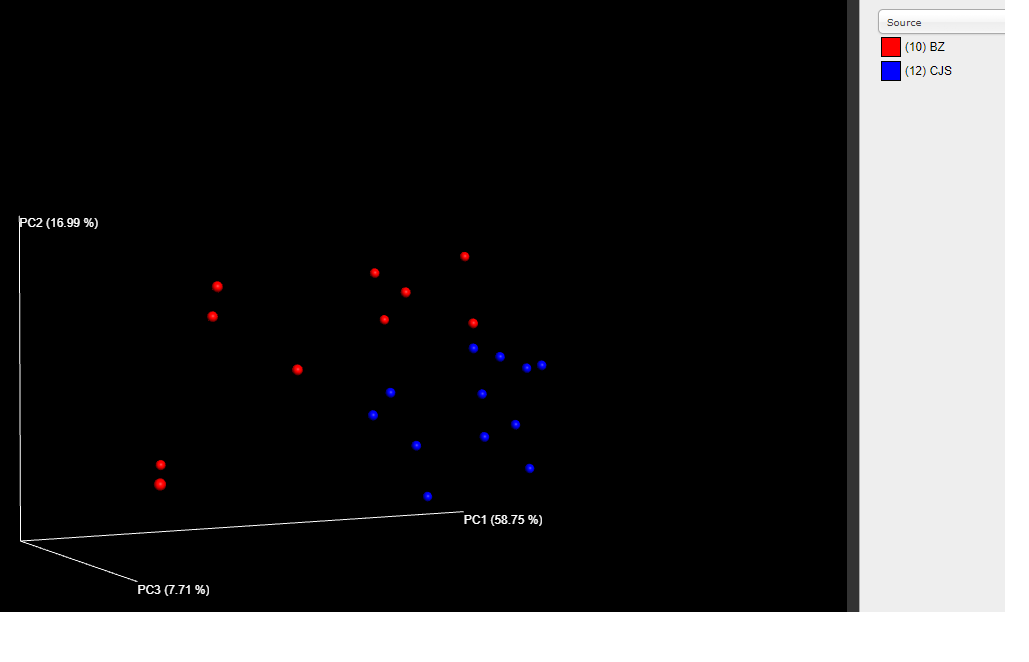
The below commands will determine if there are community-level differences between sample groupings based on weighted UniFrac distances.
Firstly, we need to align all of the DADA2 variants together. The below command uses PyNAST, which uses the Greengenes reference alignment to improve the study alignment.
align_seqs.py -i seqtab.fasta -o pynast_aligned_seqs
Importantly, whenever you use PyNAST you need to filter the alignment for low quality columns since the Greengenes alignment is based on full 16S sequences and typically study variants are of only smaller regions.
filter_alignment.py -o pynast_aligned_seqs -i pynast_aligned_seqs/seqtab_aligned.fasta
Finally we can use the below command to build a phylogenetic tree using fasttree.
make_phylogeny.py -i pynast_aligned_seqs/seqtab_aligned_pfiltered.fasta -o dada2_seqs.tre
Based on this phylogenetic tree UniFrac distances can be calculated between samples and a PCoA plot can be used to visualize these distances in multi-dimensional space.
beta_diversity_through_plots.py -m map.txt -t dada2_seqs.tre -i seqtab_tax_rarified.biom -o plots/bdiv_otu
The output plots of interest are in ```plots/bdiv_otu/

alpha_rarefaction.py -i seqtab_tax_rarified.biom -o plots/alpha_rarefaction_plot -t dada2_seqs.tre -m map.txt --min_rare_depth 25 --max_rare_depth 200 --num_steps 8

biom_to_stamp.py -m taxonomy seqtab_tax_rarified.biom > seqtab_tax_rarified.spf