{kind=link}
pyHLAMSA is a Python tool for handling Multiple Sequence Alignments (MSA) of genes with star alleles, specifically focusing on IMGT-HLA, IMGT-KIR, and CYP datasets. Key features include:
- MSA Download: Fetch MSAs for latest/specific versions.
- Gene and Allele Management: Add, delete, list, and select genes and alleles.
- Intron/Exon Operations: Manipulate specific intron/exon segments.
- Position and Region Selection: Choose targeted positions or regions.
- Alignment Operations: Add, view, concatenate, crop, and find differences.
- Genomic and Nucleotide Merging: Seamlessly merge sequences.
- Variant Operations: Calculate consensus, gather statistics, and perform variant-related tasks.
- Biopython Compatibility: Transform alignments into Biopython MultipleSeqAlignment.
- Format Conversion: Load and save MSAs in various formats (VCF, MSF, TXT, BAM, GFF).
pyHLAMSA streamlines complex gene sequence analysis, offering efficient tools for researchers.
You can simply use this package by command line.
If you want to use more powerful function, try the APIs written in below sections.
pip3 install git+https://github.com/linnil1/pyHLAMSA
# show help
pyhlamsa -h
# download kir
pyhlamsa download --family kir --db-folder tmpdir/tmp_kir_db --version 2100 tmpdir/kir --include-genes KIR2DL1 KIR2DL2
# view the msa
pyhlamsa view tmpdir/kir.KIR2DL1 --position 3-100 --include-alleles KIR2DL1*consensus KIR2DL1*063
# save the intron1+exon1 region to kir1.*
pyhlamsa view tmpdir/kir.KIR2DL1 --region intron1 exon1 --name tmpdir/kir1 --save --bam --gff --vcf --fasta-gapless --fasta-msaHLAmsa provided a simple way to read HLA data
It can automatically download and read the sequences
>>> from pyhlamsa import HLAmsa
>>> hla = HLAmsa(["A", "B"], filetype="gen",
version="3470")
>>> print(hla.list_genes())
['A', 'B']
>>> print(hla["A"])
<A gen alleles=4101 block=5UTR(301) exon1(80) intron1(130) exon2(335) intron2(273) exon3(413) intron3(666) exon4(287) intron4(119) exon5(117) intron5(444) exon6(33) intron6(143) exon7(48) intron7(170) exon8(5) 3UTR(302)>KIRmsa can also read sequences of KIR
>>> from pyhlamsa import KIRmsa
# If don't specific the genes, it will read all genes.
>>> kir = KIRmsa(ipd_folder="KIR_v2100", version="2100")
>>> print(kir.list_genes())
['KIR2DL1', 'KIR2DL2', 'KIR2DL3', 'KIR2DL4', 'KIR2DL5', 'KIR2DP1', 'KIR2DS1', 'KIR2DS2', 'KIR2DS3', 'KIR2DS4', 'KIR2DS5', 'KIR3DL1', 'KIR3DL2', 'KIR3DL3', 'KIR3DP1', 'KIR3DS1']
>>> print(kir["KIR2DL1"])
<KIR2DL1 gen alleles=173 block=5UTR(268) exon1(34) intron1(964) exon2(36) intron2(728) exon3(282) intron3(1441) exon4(300) intron4(1534) exon5(294) intron5(3157) exon6(51) intron6(4270) exon7(102) intron7(462) exon8(53) intron8(98) exon9(177) 3UTR(510)>Kind note: In our modules, exon is actually CDS, so it doesn't include UTR.
This main features give us a chance to use genomic MSA and nucleotide MSA at the same time.
The nucleotide MSA is a exon-only sequence, thus we fill the intron with E after merged.
# merge gen and nuc sequences when loading
>>> hla = HLAmsa(["A"], filetype=["gen", "nuc"],
imgt_alignment_folder="alignment_v3470")
>>> print(hla["A"])
<A gen alleles=7349 block=5UTR(301) exon1(80) intron1(130) exon2(351) intron2(273) exon3(436) intron3(666) exon4(361) intron4(119) exon5(117) intron5(444) exon6(33) intron6(143) exon7(48) intron7(170) exon8(5) 3UTR(302)>
# or manually
>>> a_gen = HLAmsa("A", filetype="gen",
>>> imgt_alignment_folder="alignment_v3470")["A"]
>>> print(a_gen)
<A gen alleles=4101 block=5UTR(301) exon1(80) intron1(130) exon2(335) intron2(273) exon3(413) intron3(666) exon4(287) intron4(119) exon5(117) intron5(444) exon6(33) intron6(143) exon7(48) intron7(170) exon8(5) 3UTR(302)>
>>> a_nuc = HLAmsa("A", filetype="nuc",
>>> imgt_alignment_folder="alignment_v3470")["A"]
>>> print(a_nuc)
<A nuc alleles=7353 block=exon1(80) exon2(351) exon3(436) exon4(361) exon5(117) exon6(33) exon7(48) exon8(5)>
>>> a_gen = a_gen.remove('A*03:437Q')
>>> print(a_gen.merge_exon(a_nuc))
<A gen alleles=7349 block=5UTR(301) exon1(80) intron1(130) exon2(351) intron2(273) exon3(436) intron3(666) exon4(361) intron4(119) exon5(117) intron5(444) exon6(33) intron6(143) exon7(48) intron7(170) exon8(5) 3UTR(302)># select exon2 and exon3
>>> exon23 = a_gen.select_exon([2,3]) # 1-base
>>> print(exon23)
<A nuc alleles=4100 block=exon2(335) exon3(413)>
# select exon2 + intron2 + exon3
>>> e2i2e3 = a_gen.select_block([3,4,5]) # 0-base
>>> print(e2i2e3)
<A alleles=4100 block=exon2(335) intron2(273) exon3(413)># select first 10 alleles
>> exon23_10 = exon23.select_allele(exon23.get_sequence_names()[:10])
>>> print(exon23_10)
<A nuc alleles=10 block=exon2(335) exon3(413)>
# print it
>>> exon23_10.print_alignment()
812 1136
| |
A*01:01:01:01 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:02N ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:03 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:04 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:05 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:06 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:07 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:08 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:09 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:10 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
# using regex to select
# "|" indicate the border of block, in this case, it's the border of exon2 and exon3
>>> exon23_1field = exon23.select_allele(r"A\*.*:01:01:01$")
>>> exon23_1field.print_alignment_diff()
812 1136
| |
A*01:01:01:01 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC.ATCCAGA TAATGTATGG CTGCGACG.. ..........
A*02:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.G------ GG-------- --------.. ..........
A*03:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.------- ---------- --------.. ..........
A*11:01:01:01 ------T-G- ---------- ---------- ---------- ---------- -| --------- --.------- ---------- --------.. ..........
A*23:01:01:01 ------A--- ----C---T- GC--T-C--- ---------- --------C- -| --------- --.C------ -G----T--- --------.. ..........
A*25:01:01:01 ------A--G ----C---T- GC--T-C--- ---------- ---------- -| --------- --.------- GG-------- --------.. ..........
A*26:01:01:01 ---------- ---------- ---------- ---------- ---------- -| --------- --.------- GG-------- --------.. ..........
A*29:01:01:01 ---------- ---------- ---------- ---------- --------C- -| --------- --.------- -G-------- ----C---.. ..........
A*30:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.------- ---------- --------.. ..........
A*32:01:01:01 ------A--G ----C---T- GC--T-C--- ---------- --------C- -| --------- --.------- -G-------- --------.. ..........
A*33:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.------- -G-------- --------.. ..........
A*34:01:01:01 ------T-G- ---------- ---------- ---------- ---------- -| --------- --.------- GG-------- --------.. ..........
A*36:01:01:01 ---------- ---------- ---------- ---------- ---------- -| --------- --.------- ---------- --------.. ..........
A*66:01:01:01 ------T-G- ---------- ---------- ---------- ---------- -| --------- --.------- GG-------- --------.. ..........
A*68:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.------- -G-------- --------.. ..........
A*69:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.G------ GG-------- --------.. ..........
A*74:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.------- -G-------- --------.. ..........
A*80:01:01:01 ---------- ---------- ---------- ---------- ---------- -| --------- --.------- ---------- --------.. ..........
# show only variantiation
>>> exon23_1field.print_snv()
Total variantion: 71
536 537 541 565 567 570 593 640 654 658 684 721
| | | | | | | | | | | |
A*01:01:01:01 | ATTTCT TCAC ATCCG| CCGGC CG CGG GGA.G | ATCGCCGTGG | .G.ACACG.C | CGTGCGGTTC GACA| AGA..A GATG| CGGGCG CCGT|
A*02:01:01:01 | ------ ---- -----| ----- -- --- ---.- | -----A---- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*03:01:01:01 | ------ ---- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*11:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*23:01:01:01 | ------ C--- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*25:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*26:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*29:01:01:01 | -----A C--- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------T ----| ---..G ----| -----A ----|
A*30:01:01:01 | ------ C--- -----| ----- A- T-- A--.- | -----A---- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*32:01:01:01 | ------ ---- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------T ----| ---..G ----| ------ ----|
A*33:01:01:01 | -----A C--- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*34:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*36:01:01:01 | ------ ---- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..- ----| ------ ----|
A*66:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*68:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*69:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*74:01:01:01 | ------ ---- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------T ----| ---..G ----| ------ ----|
A*80:01:01:01 | ------ ---- -----| ----- -- --- ---.- | -----A---- | .-.--T--.- | -----A---- ----| ---..G ----| ------ ----|# Calculate variant frequency of ( A T C G - ) per base
>>> print(exon23.calculate_frequency()[:10])
[[2, 3, 0, 4095, 0], [2, 1, 4097, 0, 0], [0, 4098, 2, 0, 0], [0, 3, 4095, 2, 0], [0, 911, 3188, 0, 1], [0, 1, 4097, 1, 1], [0, 0, 1, 0, 4099], [4097, 0, 0, 2, 1], [4, 3, 4090, 3, 0], [1, 4097, 1, 1, 0]]
# Get consensus(The largest one in frequency) among the msa
>>> exon23.get_consensus(include_gap=True)
'GCTCCC-ACTCCATGAGGTATTTCTTCACATCCGTGTCCCGGCCCGGCCGCGGGGA----GCCCCGCTTCATCGCCGTGGGC-----------------------TACGTGGACG-ACACG-CAGTTCGTGCGGTTCGACAGCGACGCCGCGAGCCAGAGGATGGAGCCG--------------------CGGGCGCCGTGGATA-GAGCAGGAGGGGCCGGAGTATTGGGACCAGGAGACACGGA-------------A-TGTGAAGGCCCACTCACAGACTGACCGAGTGGACCTGGGGACCCTGCGCGGCTACTACAACCAGAGCGAGGCCGGTTCTCACACC-ATCCAGATGATGTATGGCTGCGACG--------------TGGGG-TCGGACGGGCGCTTCCTCCGCGGGTACCAGCA---GGACGCCTACGACGGCAAGGATTAC---ATCGCCCTGAAC------------------------GAGGACCTGCGCTCTTGGACCGCGGCGGAC--------ATGGCGGCTCAGATCACCAAGCGC-AAGT----GGGAGG--CGGCCC-ATGT------------------------------------------GGCGG-AGCAGTTGAGAGCCTACCTGGAGGGCACG--------TGCGTG----GAGTGGCTCCG--CAGATA-CCTGGAGAACGGGAAGGAGACGCTGCAGC-----------------GCACGG'
# Add sequence into MSA
# include_gap=False in get_consensus will ignore the frequency of gap. i.e. choose one of ATCG
>>> a_merged = hla["A"]
>>> consensus_seq = a_merged.get_consensus(include_gap=False)
>>> a_merged.append("A*consensus", consensus_seq)
>>> a_merged.fill_imcomplete("A*consensus")
# Shrink: remove gap if all bases in the column are gap
>>> exon23_10.shrink().print_snv()
gDNA 200
|
A*01:01:01:01 AAGGCCCACT CACAGACTGA CCGAGCGAAC CTGGGGACCC TGCGCGGCTA CTACAACCAG AGCGAGGACG| GTTCTCACAC CATCCAGATA ATGTATGGCT
A*01:01:01:02N ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:03 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:04 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:05 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:06 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:07 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:08 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:09 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:10 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
# select specific column
>>> a_gen[12:100]
<A alleles=4100 block=(88)>
>>> a_gen[[12,100]].print_alignment_diff()
gDNA 0
|
A*01:01:01:01 GG
A*01:01:01:02N -G
A*01:01:01:03 GG
A*01:01:01:04 --
A*01:01:01:05 --
A*01:01:01:06 --
A*01:01:01:07 --
A*01:01:01:08 --
A*01:01:01:09 --
A*01:01:01:10 --
A*01:01:01:11 GG
A*01:01:01:12 GG
# concat
>>> print(a_gen.select_exon([2]) + a_gen.select_exon([3]))
<A nuc alleles=4100 block=exon2(335) exon3(413)>Causion: If you run merge_exon, or the msa is from filetype=['gen', 'nuc'],
You should fill the E BEFORE save it.
You can fill it by consensus_seq shown before.
-
MultipleSeqAlignment
Transfer to MultipleSeqAlignment
>>> print(a_gen.to_MultipleSeqAlignment()) Alignment with 4100 rows and 3866 columns CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...AAA A*01:01:01:01 --------------------------------------------...--- A*01:01:01:02N CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...AAA A*01:01:01:03 --------------------------------------------...--- A*01:01:01:04 --------------------------------------------...AAA A*01:01:01:05 --------------------------------------------...--- A*01:01:01:06 --------------------------------------------...AAA A*01:01:01:07 --------------------------------------------...--- A*01:01:01:08 --------------------------------------------...AAA A*01:01:01:09 --------------------------------------------...--- A*01:01:01:10 CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...--- A*01:01:01:11 CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...AAA A*01:01:01:12 --------------------------------------------...AAA A*01:01:01:13 --------------------------------------------...AAA A*01:01:01:14 --------------------------------------------...--- A*01:01:01:15 CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...--- A*01:01:01:16 CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...--- A*01:01:01:17 CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...AAA A*01:01:01:18 ... --------------------------------------------...--- A*80:07
-
list of SeqRecord
# Save as MSA SeqIO.write(a_gen.to_records(gap=True), "filename.msa.fa", "fasta") # Save as no-gapped sequences SeqIO.write(a_gen.to_records(gap=False), "filename.fa", "fasta")
-
fasta
a_gen.to_fasta("filename.msa.fa", gap=True) a_gen.to_fasta("filename.fa", gap=False)
-
bam
a_gen.to_bam("filename.bam")
-
gff
a_gen.to_gff("filename.gff")
After save the MSA as bam and gff, you can show the alignments on IGV
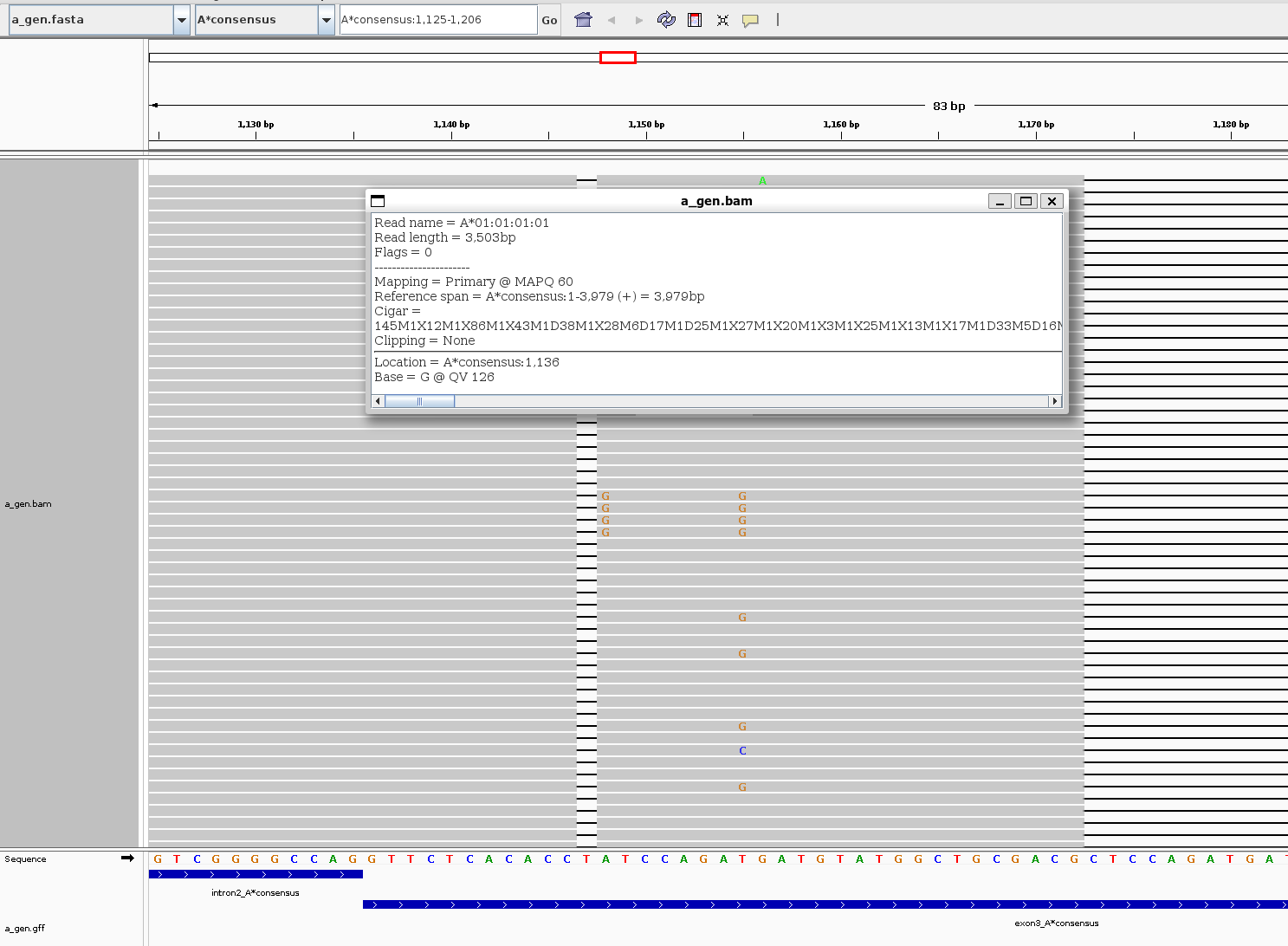
-
vcf
a_gen.to_vcf("filename.vcf.gz")
-
IMGT MSA format (xx_gen.txt)
a_gen.to_imgt_alignment("A_gen.txt") a_gen.to_imgt_alignment("A_nuc.txt", seq_type="nuc")
-
save/load
Save our model in fasta and json, where json contains block, index information
a_gen.save_msa("a_gen.fa", "a_gen.json") a_gen = Genemsa.load_msa("a_gen.fa", "a_gen.json")
-
load msa from other format
pyHLAMSA only support reading from
MultipleSeqAlignment, which is very useful object, can be generate by reading MSA byBio.AlignIO.Checkout https://biopython.org/wiki/AlignIO#file-formats for format supporting.
For example
from pyhlamsa import Genemsa msa = Genemsa.from_MultipleSeqAlignment(AlignIO.read(your_data_path, your_data_format))
- Testing
- Main function
- exon-only sequence handling
- Reading from file
- Some useful function:
copy,remove,get_sequence_names,__len__,size,split,concat - Cannot handle pseudo exon
- merge blocks and labels
- Fix KIR when merge gen and nuc
- Use index to trace orignal position
- CYP
- Set reference
- Download latest version of IMGT or IPD
- Remove seqtype
- Split code
- save to VCF
- Add command line usage
- Remove msaio
- Rewrite gene Mixins (Too complicated)
- Fix selction and removeing allele by regex
- Change to ACGT order
- Rename: split -> split_block, remove -> remove_allele
- CDS != exon, (rename it?)
- Rename: pyHLAMSA -> py_star_msa (MAYBE, becuase the project is originally written for HLA)
- python3.9
- biopython
- pysam
- wget
- git
pip3 install git+https://github.com/linnil1/pyHLAMSA
# or
git clone https://github.com/linnil1/pyHLAMSA
pip3 install -e pyHLAMSASteps:
- Download fasta from https://www.pharmvar.org/download
- unzip to
./pharmvar-5.1.10 - Read it by pyhlamsa
# 4. Read it
from pyhlamsa import CYPmsa
cyp = CYPmsa(pharmvar_folder="./pharmvar-5.1.10")
# 5. Test it
>>> print(cyp['CYP26A1'].format_variantion_base())
6407 6448 8142
| | |
CYP26A1*1.001 GCGAGCGCGG | ATGTTCCGAA | TCGGGTGTGT
CYP26A1*3.001 ---------- | -----A---- | ----------
CYP26A1*2.001 -----A---- | ---------- | ----------
CYP26A1*4.001 ---------- | ---------- | -----C----pip3 install mkdocs-material mkdocstrings[python-lagacy]==0.18
mkdocs serveI use python-lagacy 0.18 because inherited_members is not support now
pip3 install -e .
pip3 install pytest black mypy
pytest
black pyhlamsa
mypy pyhlamsa- CYP: 5.2.2
- HLA: 3.49.0
- KIR: 2.10.0 (The DB in 2.11.0 contains bugs in KIR2DL4/5)
Why not inherit Bio.AlignIO.MultipleSeqAlignment?
The class does't support lot of functions than I expected.
And it's tidious to overwrite most of the functions to maintain our blocks information
Why not use numpy to save the sequence?
Performance issue is not my bottle-neck yet.
- HLA Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE: IPD-IMGT/HLA Database. Nucleic Acids Research (2020), 48:D948-55
- IMGT Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE IPD-IMGT/HLA Database Nucleic Acids Research (2020) 48:D948-55
- CYP(PharmVar) Pharmacogene Variation Consortium (PharmVar) at www.PharmVar.org using the following references: Gaedigk et al. 2018, CPT 103(3):399-401 (PMID 29134625); Gaedigk et al. 2020, CPT 107(1):43-46 (PMID 31758698) and Gaedigk et al. 2021, CPT 110(3):542-545 (PMID 34091888)
- This github
See [https://linnil1.github.io/pyHLAMSA](See https://linnil1.github.io/pyHLAMSA)
::: pyhlamsa.gene.genemsa ::: pyhlamsa.gene_family.hla ::: pyhlamsa.gene_family.hla_ex ::: pyhlamsa.gene_family.kir ::: pyhlamsa.gene_family.cyp