

![]()

💫 Graph-based foundation model for spatial transcriptomics data
Novae is a deep learning model for spatial domain assignments of spatial transcriptomics data (at both single-cell or spot resolution). It works across multiple gene panels, tissues, and technologies. Novae offers several additional features, including: (i) native batch-effect correction, (ii) analysis of spatially variable genes and pathways, and (iii) architecture analysis of tissue slides.
Note
Novae was developed by the authors of sopa and is part of the scverse ecosystem. Read our article here.
Check Novae's documentation to get started. It contains installation explanations, API details, and tutorials.
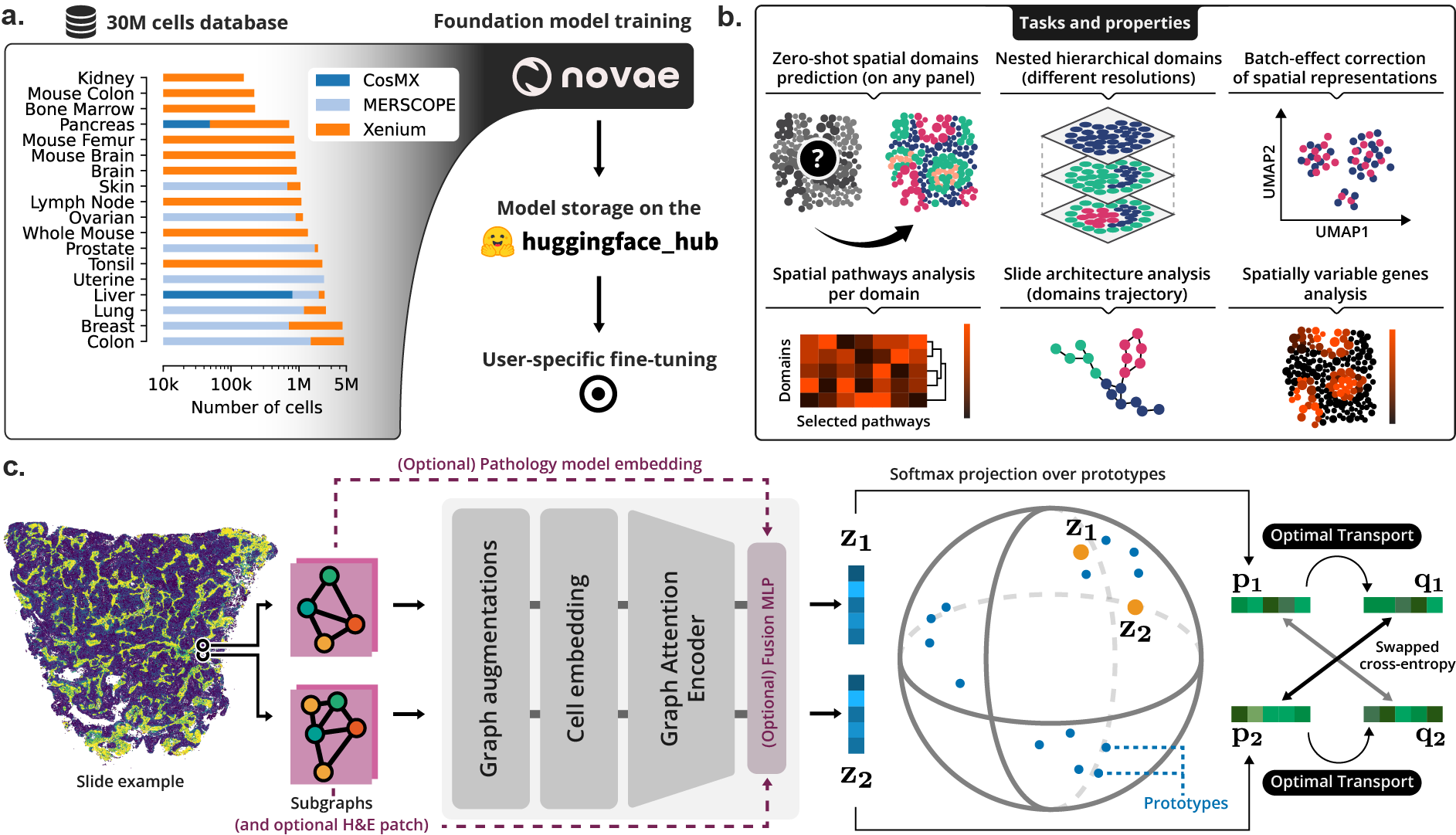
(a) Novae was trained on a large dataset, and is shared on Hugging Face Hub. (b) Illustration of the main tasks and properties of Novae. (c) Illustration of the method behind Novae (self-supervision on graphs, adapted from SwAV).
novae can be installed from PyPI on all OS, for any Python version >=3.11 and <3.14 (3.14 support coming soon).
pip install novae
Note
See this installation section for more details about extras and other installations modes.
Here is a minimal usage example. For more details, refer to the documentation.
import novae
# compute cell neighbors
novae.spatial_neighbors(adata)
# load a pre-trained model
model = novae.Novae.from_pretrained("MICS-Lab/novae-human-0")
# compute spatial domains
model.compute_representations(adata, zero_shot=True)
model.assign_domains(adata)Our article is published in Nature Methods. You can cite Novae as below:
Blampey, Q., Benkirane, H., Bercovici, N. et al. Novae: a graph-based foundation model for spatial transcriptomics data.
Nat Methods (2025). https://doi.org/10.1038/s41592-025-02899-6